1. Introduction
In heterogeneous catalytic reactions, the Au nanoparticles (NPs), as the active species with superior catalytic activities, have been attracted considerable interests in recent decades. They are active in a great deal of oxidation and reduction reactions, especially in the oxidation of CO [
1]. Most intensely studied are not only Au/CeO
2 catalysts, which have emerged as one of the best candidates for the low temperature CO oxidation [
2,
3], but also other catalysts such as Au/HAP [
4], Au/Fe
2O
3 [
5], Au/TiO
2 [
6] and Au/ZrO
2 [
7] have been investigated. However, the major problem of supported Au catalysts is the quick deactivation due to the low thermal stability during the catalytic reactions, thus, hindering their practical application. Au NPs are thermodynamically unstable and tend to be easily sintered when calcined at elevated temperatures or a long period of time. Since then, the active supported Au catalysts with the sintering-resistant property have been covered widely by numerous studies. As a result, it has been confirmed that the catalytic activity strongly depends on the change of Au particle size [
8], the appropriate choice of support materials (oxides/non-oxides) [
9], the formation of carbonates adsorbed on the active sites [
10] and the metal-support interactions [
11].
Several strategies have been developed to stabilize Au NPs for CO oxidation processes. Chen’s group reported that the enhancing of catalytic activity and the various carbonate species are contributed by the different small Au particle sizes [
8]. Putla et al. introduced various dopants to Au/CeO
2, and found that the appropriate support materials is able to improve the catalytic properties of Au based catalysts [
12]. Qiao et al. demonstrated a classical strong metal–support interaction (SMSI) for Au/TiO
2, which markedly depended on the reversible encapsulation of Au NPs by TiO
2 support following high-temperature redox pretreatments [
13]. Park’s group performed an oxidation atmosphere pretreatment on Au/TiO
2 catalysts, and found that the oxidation pretreatment enhanced SMSI with the improved activity of CO oxidation [
14]. However, the structural effects associated with the pretreatment process are not well understood. Because the Au NPs calcined at high temperatures are easily agglomerated after pretreatments [
15], there are few efficient methods to improve the thermal stability of Au NPs with reduction conditions.
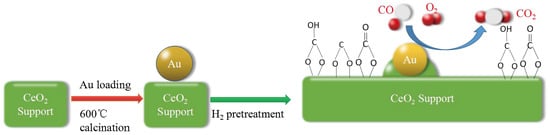
Here, inspired by previous research results, H2-reducing pretreatment method was used to fabricate SMSI in the Au-CeO2 catalysts further to trace whether the Au NPs can be stabilized while holding their high activity. So, in our study, the Au-CeO2 spheres calcined at 600 °C for various times, followed by pretreated upon H2 atmosphere at 200 °C. These products were characterized by X-ray diffraction (XRD), N2 adsorption–desorption, high-resolution transmission electron microscope (HRTEM), in-situ diffuse reflectance infrared Fourier transform spectroscopy (in-situ DRIFTS) and X-ray photoelectron spectra (XPS) to elucidate the effect of H2 pretreatments on the catalytic performance of CO oxidation. After H2 pretreatment, the introduction of SMSI in the Au-CeO2 catalysts could provide the enhanced activity and thermal stability until the calcination times increased to 12 h. Thus, the H2 pretreatment makes the Au NPs more resistant to sintering at high temperatures, which may be extended to other supported noble metal catalysts.
3. Results and Discussion
Catalytic CO oxidation is used as a probe reaction to investigate the relations between the pretreatment conditions and the catalytic properties of the Au-CeO
2 catalysts.
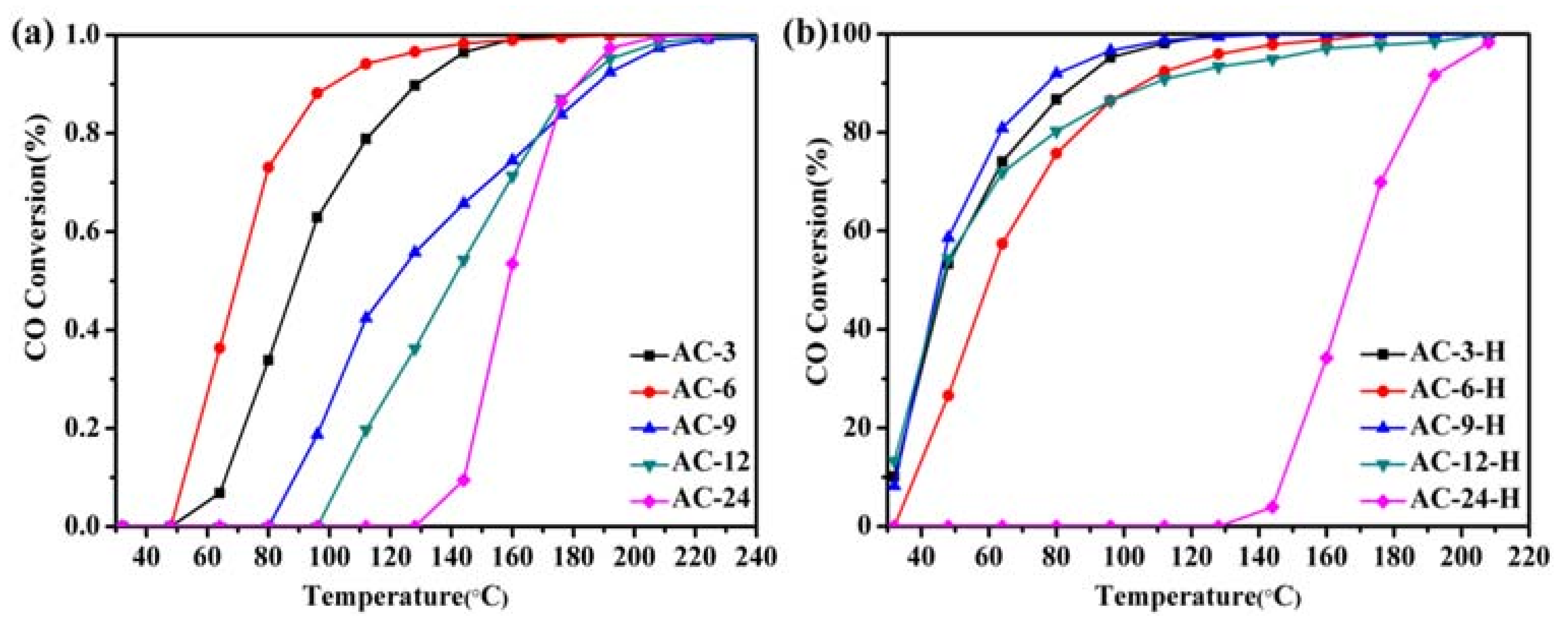
Figure 1 describes the CO conversion profiles of the various Au-CeO
2 samples calcined at 600 °C for 3–24 h. The T
50 and T
90 (temperatures for 50% and 90% CO conversion) of different samples were summarized in (
Table S1) to compare the activity of the catalysts. For the Au-CeO
2 samples calcined at different prolonged times without H
2 pretreatments, the CO oxidation activities decrease in the order: AC-6 > AC-3 > AC-9 > AC-12 > AC-24 (
Figure 1a). Obviously, the deactivation could occur in the Au-CeO
2 samples without H
2 pretreatment as the prolonged calcination time. Before the catalytic process, introducing the H
2 pretreatments to the samples causes a significant enhancement of the catalytic activity (
Figure 1b). The catalytic activity of AC-3 with uncovered Au NPs is significantly lower than that of AC-3-H with partially covered Au NPs. However, the AC-6-H sample (T
50 = 60 °C, T
90 = 106 °C) has a similar catalytic activity on AC-6 sample (T
50 = 70 °C, T
90 = 101 °C), indicating the active site of the AC-6 sample after H
2 pretreatment is blocked. When the calcination time arises to 12 h, the samples with H
2 pretreatment also provides stable catalytic performance (T
50 = 46 °C, T
90 = 111 °C). Especially, the AC-9-H sample has better catalytic activities, which gives the T
50 of 45 °C and T
90 of 77 °C.
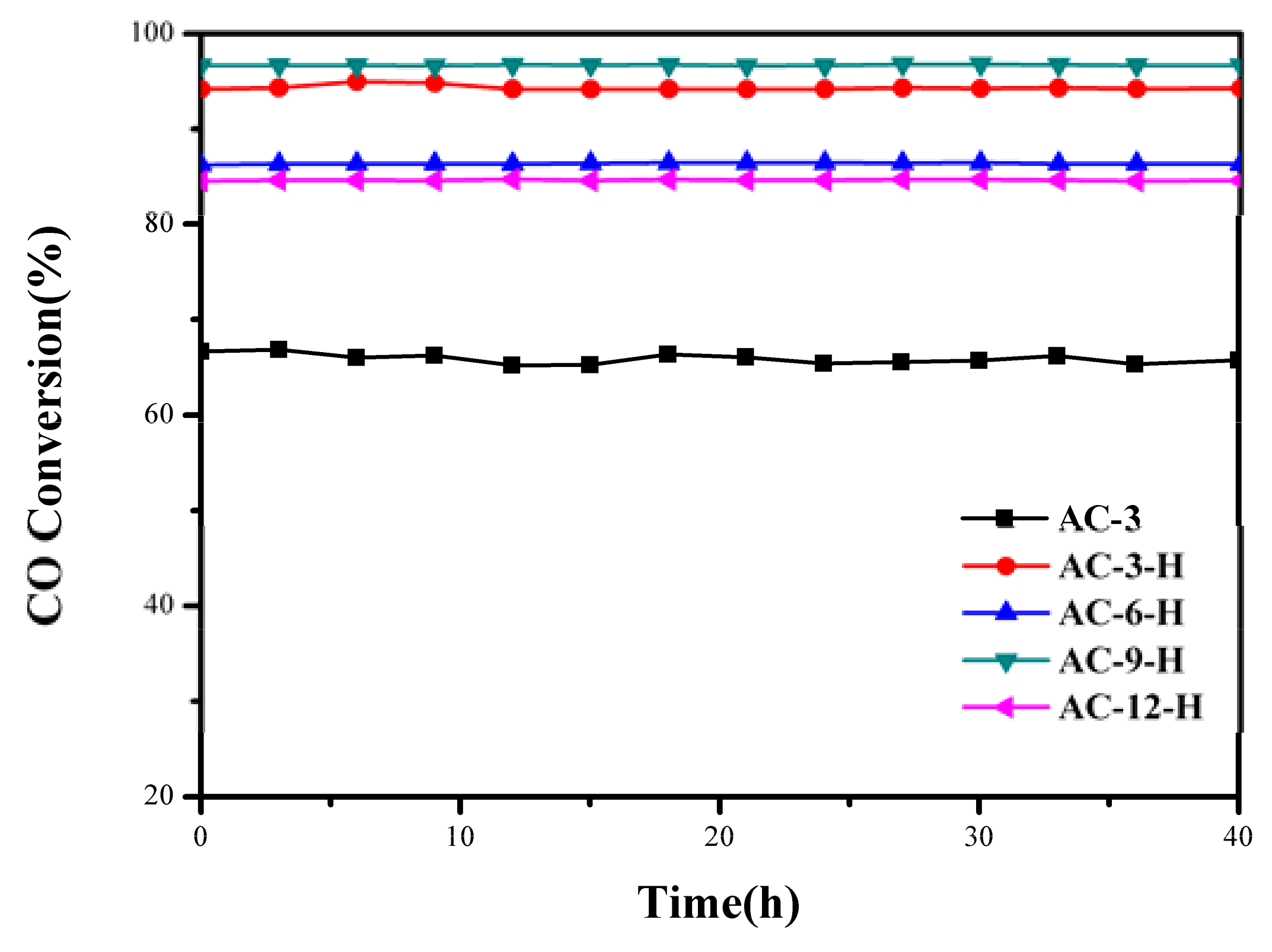
To better evaluate the thermal sintering performances, the temporal evolution profiles of the CO oxidation over the Au-CeO
2 samples with and without H
2 pretreatments (reaction at 100 °C for 48 h) are illustrated in
Figure 2. There was no obvious deactivation in CO conversion, demonstrating its sintering-resistant catalytic performance. Overall, it can be found that H
2 pretreatment could obtain thermally stable Au-CeO
2 samples with a higher activity in comparison to samples without H
2 pretreatments.
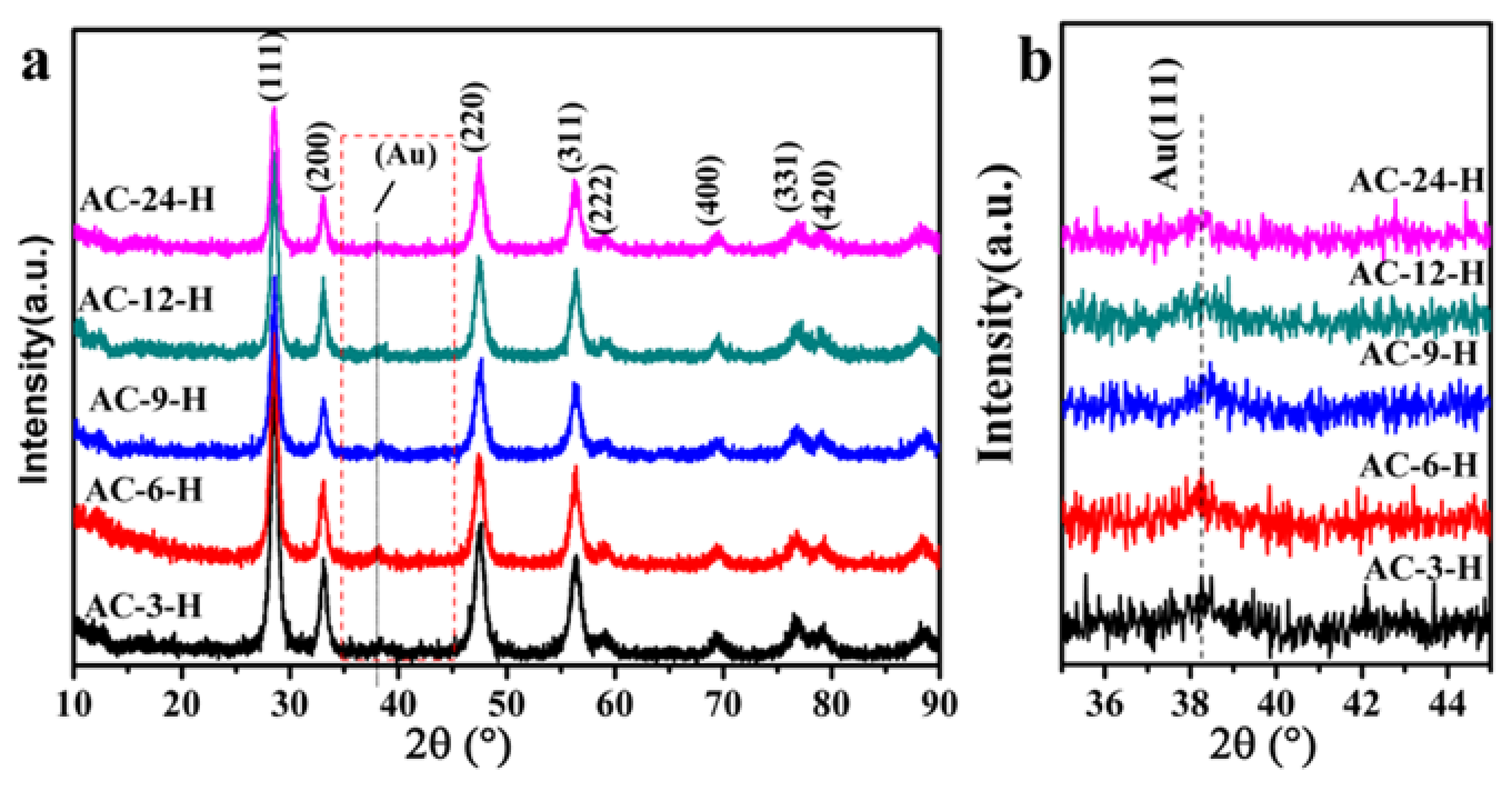
The XRD profiles of different Au-CeO
2 samples are shown in
Figure 3 (with H
2 pretreatment) and
Figure S1 (without H
2 pretreatment). The reference pure CeO
2 sample in
Figure S1 showed weak diffraction peaks with a lower average crystallite size of 5.0 nm (summarized in
Table S2). All Au-CeO
2 samples with or without H
2 pretreatment clearly show obvious diffraction peaks at 2θ value of 28.5° (111), 33.1° (200), 47.4° (220), 56.3° (311), 59.0° (222), 69.4° (400), 79.1° (331) and 88.4° (420), which are the fluorite-type cubic structure of CeO
2 [
16]. Besides, the diffuse diffraction peaks of Au (111) located at 38.2°, suggesting the characteristic of Au NPs [
17].
In order to know the influence of the calcination time on the textural properties of CeO
2, average crystallite sizes were calculated from the Debye–Scherrer equation and specific surface areas were determined from N
2 adsorption-desorption test (
Table 1). It clearly shows that the specific surface area of Au-CeO
2 samples is about 30–60 m
2/g with pore volume of 0.14–0.20 cm
3/g (
Table S1). For the unpretreated Au-CeO
2 samples, the prolonged calcination time is accompanied by the increase of CeO
2 average crystallite size (
Table S2). After H
2 pretreatment, the CeO
2 average crystallite size for all Au-CeO
2 samples is decreased, implying that H
2 pretreatment may restrain the size growth of CeO
2. There is a decrease trend in surface area with the prolonged calcination time which correlates well with the increase of average crystallite size except the AC-9-H sample. The meticulous correlation of crystallite size and surface area of Au-CeO
2 samples reveals the unusual crystallite size and surface area of AC-9-H sample. H
2 pretreatment causes an obvious variation of average crystallite size for Au-CeO
2 samples, which may lead to the unusual surface area for AC-9-H sample.
Transmission electron microscope (TEM) is further used to identify microscopic structures of Au-CeO
2 with or without H
2 pretreatment. As shown in
Figure 4, highly dispersed Au NPs are successfully attached on the CeO
2 hollow nanospheres. For all Au-CeO
2 samples, the CeO
2 nanospheres have a particle size of ~120 nm (
Table S3). With the increase of calcination time at 600 °C, there is no obvious agglomeration for CeO
2 nanospheres. Significantly, it is clearly found that the sizes of Au NPs increase with the prolonged calcination time following in the order: AC-3-H (9.8 nm) < AC-6-H (10.1 nm) < AC-9-H (10.2 nm) < AC-12-H (12.0 nm) < AC-24-H (13.0 nm) (
Table S3).
As can be seen in
Figure 5, the typical HRTEM micrographs of the single Au-CeO
2 nanosphere reveal the presence of Au particles, in contact with the CeO
2 surfaces. The fringe period of ~0.312 nm and ~0.271 nm is respectively in agreement with the (111) and (200) lattice spacing of CeO
2, the lattice fringes of ~0.236 nm and ~0.204 nm are consistent with the (111) and (200) crystal plane of metallic Au. As shown in
Figure 5a, Au NPs on the AC-3 sample without H
2 pretreatment were naked. After H
2 pretreatment, several important characters can be distinguished for the different samples in
Figure 5b–f: (i) the naked and covered Au NPs coexist for AC-3-H, AC-9-H, AC-12-H and AC-24-H samples; (ii) whereas for AC-6-H sample, the coverage increased rapidly and the Au NPs are almost completely covered. Interestingly, AC-6-H is the only sample with Au NPs completely covered, which may be due to the increase of Au NPs and CeO
2 crystallite size with the prolonged calcination time and/or the reduction of H
2 pretreatment. The coverage of oxides support on metal NPs is a typical feature of SMSI phenomenon [
13]. As a result, the coverage of CeO
2 support on the Au NPs indicates the formation of SMSI in our Au-CeO
2 catalysts with H
2 pretreatments.
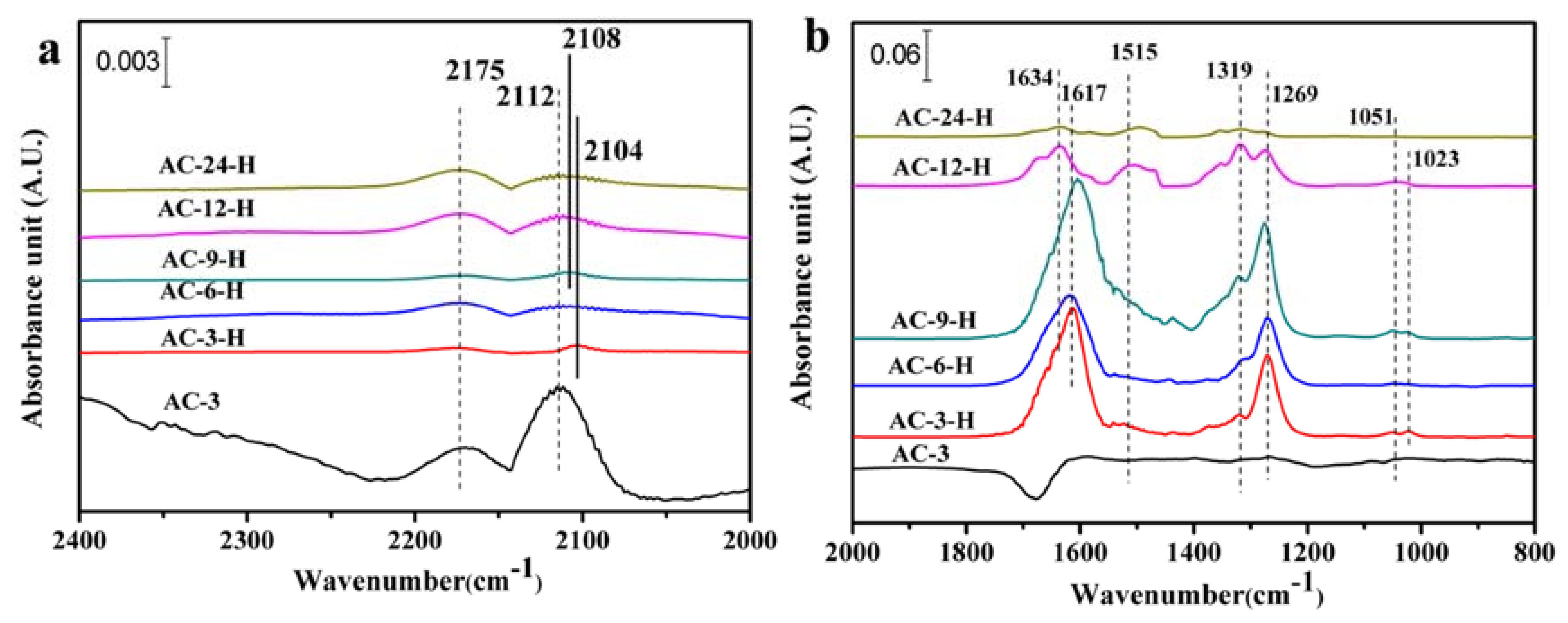
The SMSI generally results in fewer CO adsorption sites on Au NPs, because the CO adsorption sites are covered by the support layer [
17]. The in-situ DRIFTS measurements of CO adsorption with a suitable probe molecule were used to explore the CO adsorption change and/or electron transfers of the Au-CeO
2 catalysts. As shown in
Figure 6a, two bands are detected on the 2168–2177 cm
−1 and 2104–2112 cm
−1 for all the samples. The former is assigned to gaseous CO [
18], and the later can vest in CO at the metallic Au (CO-Au
0) [
19]. Obviously, the unpretreated AC-3 sample exhibits the highest peak intensities for the two bands with CO-Au
0 centered at 2112 cm
−1. Additionally, for the other unpretreated Au-CeO
2 samples, the CO-Au
0 band is also detected at around 2112 cm
−1 (shown in
Figure S2a). After H
2 pretreatment, the CO-Au
0 band for AC-3-H sample is blue shifted to 2104 cm
−1 with the peak intensity largely decreased. As the calcination time increased, there is a blue shift for CO-Au
0 absorbed species on Au-CeO
2 samples, giving 2108 cm
−1 over AC-9-H, and 2112 cm
−1 over AC-6-H, AC-12-H and AC-24-H. After H
2 pretreatment, the CO-Au
0 band on Au-CeO
2 samples is blue shifted, indicating more electropositive Au species can be formed on Au-CeO
2 with H
2 pretreatment. This blue-shift of the CO adsorption suggests the electron transfer from the CeO
2 support to Au NPs [
20]. Besides, the AC-6-H sample gives a lower intensity at the CO-Au
0 band. According to the previous reports [
17], the decreased CO adsorption is related to the loss in CO adsorption sites mainly deriving from the partial cover of Au NPs by CeO
2 supports. In our work, the H
2 pretreatment could be a powerful approach to alter the electron interactions between Au NPs and CeO
2.
The in-situ DRIFT spectra of CO adsorption in the region of 2000–800 cm
−1 (
Figure 6b and
Figure S2) shows bands corresponding to various carbonate surface substances, which are basically chemisorbed on CeO
2. The presence of various carbonate species is summarized in
Table S4. Further, Several important carbonate species can be distinguished for the Au-CeO
2 catalysts: tridentate carbonates (bands at 1048–1073 cm
−1 and 1266–1276 cm
−1 and 1460–1550 cm
−1), bidentate carbonates (bands at 1014–1028 cm
−1 and ~1319 cm
−1) and bicarbonate species (bands at 1600–1616 cm
−1 and 1618–1638 cm
−1) [
21]. The negative bands at 1600–1700 cm
−1 in
Figure 6b and
Figure S2b may be ascribed to the adsorption of water vapor on the sample before IR tests. However, only traces amount of tridentate carbonates and bidentate carbonates are formed on AC-3 sample. The carbonate surface species are more easily formed after H
2 pretreatment. It can be noted from HRTEM results that the H
2 pretreatment leads to a closer connection between Au NPs and CeO
2 which facilitates CO oxidation at low temperatures [
9]. Thus, the produced CO
2 may be adsorbed on the ceria surfaces forming the large amounts of carbonates. When the Au-CeO
2 samples are exposed to CO, the CO molecule can be adsorbed on the active Au NPs, forming CO-Au
0 species. The lattice oxygen atoms on the surface of CeO
2 are more reactive and can react with the adsorbed CO molecule, producing carbonates, biocarbonates species and oxygen vacancy. Finally, the surface carbonates species decomposed to CO
2, with the remaining oxygen atom filling the vacancy. In fact, the proposed reaction mechanisms have already been widely accepted by other researches [
22].
For the Au-CeO
2 samples with H
2 pretreatments, the band intensity of these carbonate species decreases in the order: AC-9-H > AC-3-H > AC-6-H > AC-12-H > AC-24-H. Among all samples with H
2 pretreatments, AC-9-H and AC-3-H samples exhibit relatively larger band intensity of carbonate species, which may be ascribed to their higher surface areas compared with other samples. Significantly, bicarbonate species have been detected for all samples and account for the vast majority of these carbonates species. Moreover, when the calcination time increased to 24 h, the peak intensity of the bicarbonates species decreased sharply. It is well known [
18,
23] that the surface bicarbonates are less thermally stable than carbonates species, which facilitates faster desorption of CO
2, allowing the surface effective for further chemisorption. Therefore, the presence of bicarbonates on the Au-CeO
2 under H
2 pretreatments can accelerate CO oxidation.
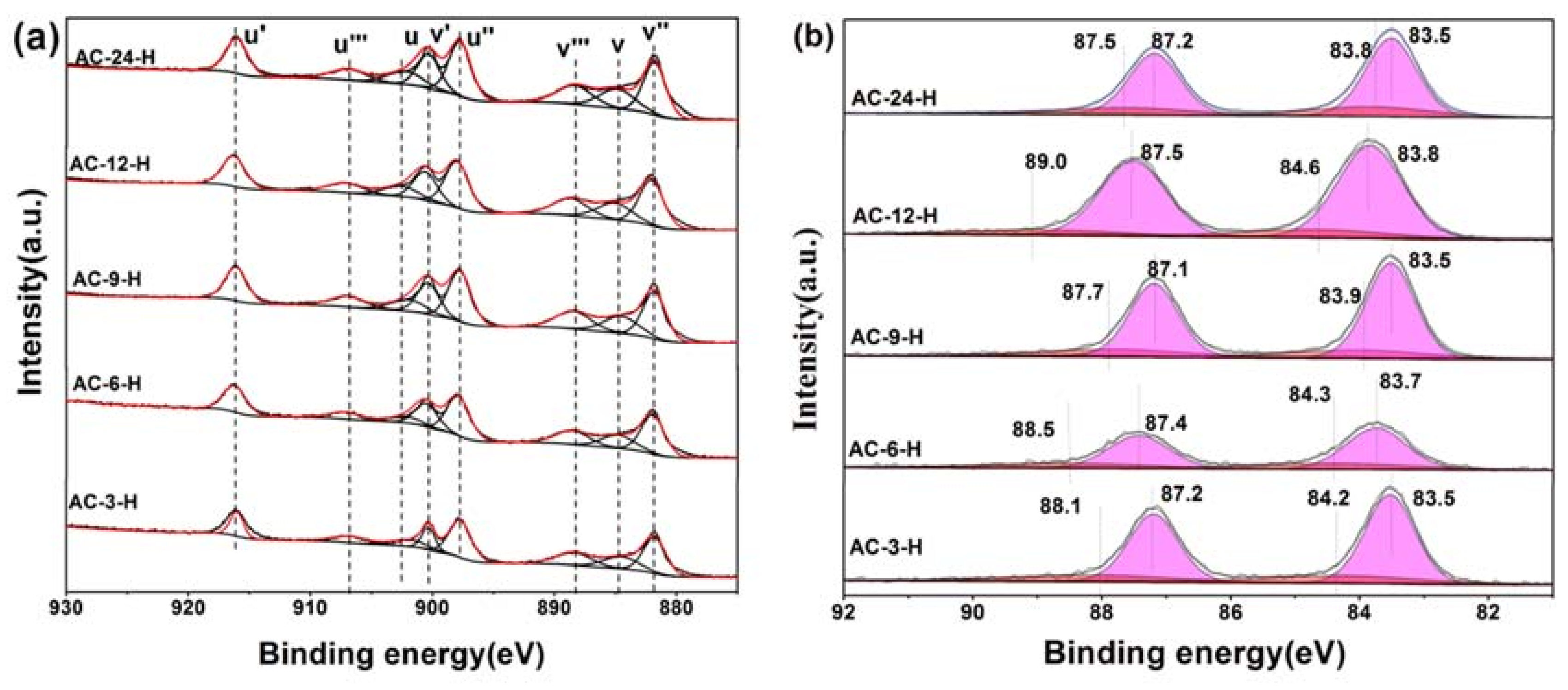
X-ray photoelectron spectroscopy (XPS) experiments were conducted in order to confirm whether there is a change in the valence states of the elements in the Au-CeO
2 samples with or without H
2 pretreatment.
Figure 7 shows the XPS spectra of Au 4f and Ce 3d for different catalysts with H
2 pretreatment, while
Figure S3 shows the XPS spectra of AC-3 without H
2 pretreatment. In the Ce 3d region, the peaks marked as u’ (916.0 eV), v’ (900.3 eV), u’’ (898.0 eV), v’’’ (882.0 eV), u’’’ (907.0 eV) and v’’’ (888.5 eV) correspond to the Ce
4+ state, whereas that denoted as u (902.1 eV) and v (884.5 eV) are assigned to Ce
3+ [
24]. In
Table S5, the results of the primary binding energies Ce 3d achieved by the XPS quantitative analysis, are reported for the different samples. There is no difference in the Ce 3d spectra of these samples except AC-6-H, indicating their similar electronic properties. Clearly, the over encapsulation of Au NPs for AC-6-H may result in a lower content of Ce
3+.
In the Au 4f region, for quantitative evaluation, the Au 4f peaks with binding energies (BEs) were determined to be two different states. After curve fitting, the Au 4f peaks with binding energies of about 84.3 and 87.8 eV in AC-3 sample are attributed to the presence of Au
0, while two weak BE peaks at 88.4 and 84.6 eV indicate the existence of the Au
δ+ species [
21]. After H
2 pretreatment, a visible shift of BE to 83.5 eV–83.8 eV was observed on Ae-CeO
2 samples, implying the Au NPs supported on CeO
2 become electron-rich [
11]. As a consequence of this electron-rich effect, the atomic ratio of the Au
δ+ species for Au-CeO
2 after H
2 pretreatment decreases rapidly (
Table S6). Overall, the electron transfers between Au NPs and CeO
2 are created to make their interface more closely connected, leading to electron-rich Au after H
2 pretreatment [
4]. According to the in-situ DRIFTS, XPS and HRTEM results, the particular coverage of Au nanoparticles by CeO
2, the electron transfers and the CO adsorption changes are identical to those characteristics in SMSI.
There are many efficient strategies for stabilizing Au NPs on various supports, such as the synthetic of yolk–shell nanoparticles [
25], the coverage of Au NPs substrate coatings and the surface-modified of supports before Au NPs loading [
26]. After H
2 pretreatment, our CO oxidation results imply that the Au-CeO
2 catalysts provide the enhanced activity and thermal stability until the elevated calcination times increased to 12 h. This is due to the occurrence of SMSI and the presence of bicarbonates. After H
2 pretreatment, the construction of SMSI can be proposed for Au-CeO
2 catalysts where the particular coverage of Au nanoparticles by CeO
2, the electron transfers and the CO adsorption changes are in good agreement with those in classic SMSI. As for the non-linearity in the catalytic performance, the possible reason can be ascribed to the different coverage degree of Au NPs on the CeO
2 support and the size variation of Au NPs. The partially exposed Au NPs for AC-3-H and AC-9-H, which are in direct contact with CeO
2, the show the better catalytic activities for CO oxidation. On the contrary, the over-encapsulation of Au NPs on AC-6-H can suppress the active sites, resulting in relatively low activities. Moreover, the AC-24-H sample has poorer catalytic activities than others due to the relatively larger Au NPs. Besides, the surface bicarbonates would favor a faster desorption of CO
2, through which the CO oxidation is accelerated. Moreover, the AC-24-H sample has poorer catalytic activities than others due to the relatively larger Au NPs and less bicarbonates surface species, which are still an important factor to limit the catalytic activity.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}