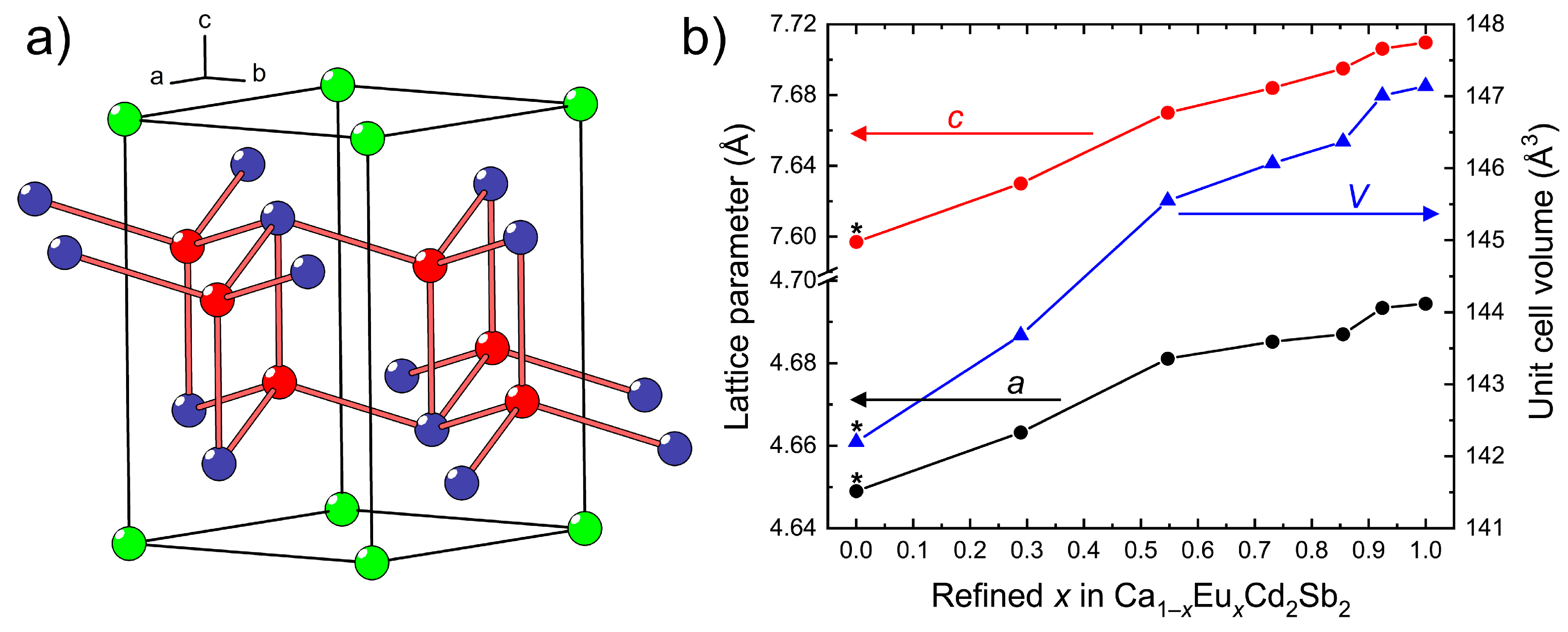
3.1. Crystal Structure of the Ca1–xEuxCd2Sb2 Solid Solution
The Ca
1−xEu
xCd
2Sb
2 solid solution crystallizes in the space group,
Pm1, and adopts the CaAl
2Si
2 structure type (Pearson code
hP5), which can be also viewed as a ternary derivative of the La
2O
3 type (
Figure 1a). Atomic coordinates and equivalent displacement parameters for Ca
1−xEu
xCd
2Sb
2 with
x = 0.29(1) are presented in
Table 2. Crystallographic data for the other studied compositions are given in the Supporting Information.
The crystal structure accommodates two-dimensional slabs of corner- and edge-sharing CdSb
4 tetrahedra alternating with hexagonal layers of
AE atoms, where
AE = Ca + Eu. The
AE atoms are six-fold coordinated by the adjacent Sb atoms, forming almost perfectly regular octahedra. Assignment of the formal charges to the atoms in the structure yields an electron-balanced composition according to the notation (
AE2+)(Cd
2+)
2(Sb
3−)
2. The end members of these series with
x = 0 (CaCd
2Sb
2) and
x = 1 (EuCd
2Sb
2) are also known and crystallize in the same structure [
26]. In addition, various other pnictides with the
AETM2Pn2 composition (
AE = alkaline-earth metal, Eu, or Yb;
TM = transition metal;
Pn = pnictogen) adopt this type [
27,
28,
29]. Many of these phases have been studied as potential thermoelectric materials owing to their narrow-gap semiconducting properties [
30,
31,
32,
33,
34,
35,
36].
In the Ca
1−xEu
xCd
2Sb
2 series, the lattice parameters and the unit cell volume gradually increase on going from the Eu-poorer to the Eu-richer compositions (
Figure 1b), in accordance with a larger ionic size of Eu
2+ in comparison with Ca
2+ (for the same coordination number 6 for instance,
r(Ca
2+) = 1.00 Å,
r(Eu
2+) = 1.17 Å [
37]). The Vegard’s law is not strictly observed, i.e., the change of the unit cell dimensions is not linear, suggesting that local ordering may be taking place. Analysis of the single-crystal diffraction data gives no indication of superstructure reflections, thus confirming the preservation of the long-range CaAl
2Si
2 structure.
In line with the increasing average size of the
AE2+ cation in
AECd
2Sb
2 (
AE = Ca + Eu), the
AE–Sb contacts become progressively longer upon increasing the level of Eu substitution (
Table 3). At the same time, the Cd–Sb bonds remain virtually unaffected. Another noteworthy structural change occurring as the Eu concentration increases concerns the Sb–
AE–Sb bond angles, and the trend is suggestive of adopting a more regular octahedral environment of the
AE atoms.
3.2. Crystal and Electronic Structure of the Ca2−xEuxCdSb2 Solid Solution (x ≈ 0.6)
A crystal of the new solid solution with the composition, Ca
2−xEu
xCdSb
2 (
x ≈ 0.6), was discovered as a by-product in the sample containing Ca
1−xEu
xCd
2Sb
2 as the main phase. The crystal structure of Ca
2−xEu
xCdSb
2 belongs to the Yb
2CdSb
2 type (Pearson code
oS20, space group
Cmc2
1), in contrast to the parent ternary phase, Ca
2CdSb
2, crystallizing in its own type (Pearson code
oP20, space group
Pnma) [
38]. The two prototypic structures possess no homoatomic Sb–Sb bonds and can therefore be rationalized within the Zintl concept as (
AE2+)
2(Cd
2+)(Sb
3−)
2, where
AE stands for Yb or Ca. The main difference between the two types stems from non-identical stacking of the layers built up of corner-sharing CdSb
4 tetrahedra. Whereas in the non-centrosymmetric Yb
2CdSb
2, the adjacent Cd-Sb layers are related only by translation, in Ca
2CdSb
2, similar layers alternate along the stacking direction with their crystallographically inverted counterparts.
From another point of view, the structures of Yb
2CdSb
2 and Ca
2CdSb
2 can be understood as two different ways of occupying tetrahedral voids in otherwise isostructural Yb–Sb or Ca–Sb substructures [
38]. As local atomic environments in both types appear to be almost identical, subtle electronic or size differences must be responsible for stabilization of one or another structure. As a matter of fact, several pnictides with the “2-1-2” composition have been found to crystallize in the Yb
2CdSb
2 structure [
39,
40], whereas Ca
2CdSb
2 remains the only representative of its type. In this regard, it is important to mention that the Zn-variants with the same composition are totally different (defect ZrBeSi-type structure) [
41], and the Mn-based “2-1-2” analogs adopt their own structure [
42,
43]. Within this context, it also is instructive to recall that a ternary compound, “Eu
2CdSb
2”, with either a Ca
2CdSb
2 or Yb
2CdSb
2 structure type, is not known to exist (to date). Apparently, the Ca
2CdSb
2 structure becomes destabilized upon isovalent substitution of Ca with Eu, yielding a solid solution with the non-centrosymmetric Yb
2CdSb
2 atomic arrangement (
Figure 2a). Further optimization of the synthetic procedure will be required to systematically study the structural evolution of the “2-1-2” phase in a wider compositional range and to find the critical Eu content necessary to disrupt the original Ca
2CdSb
2 structure.
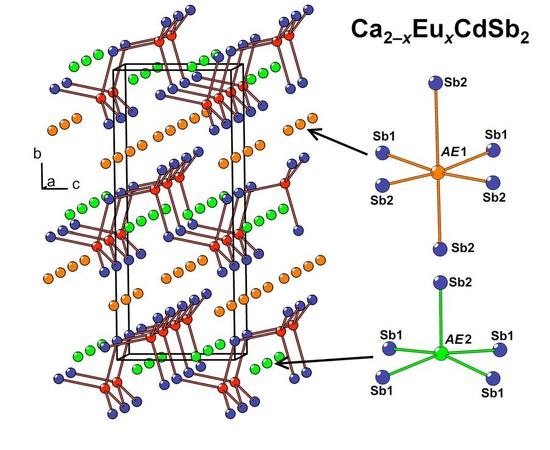
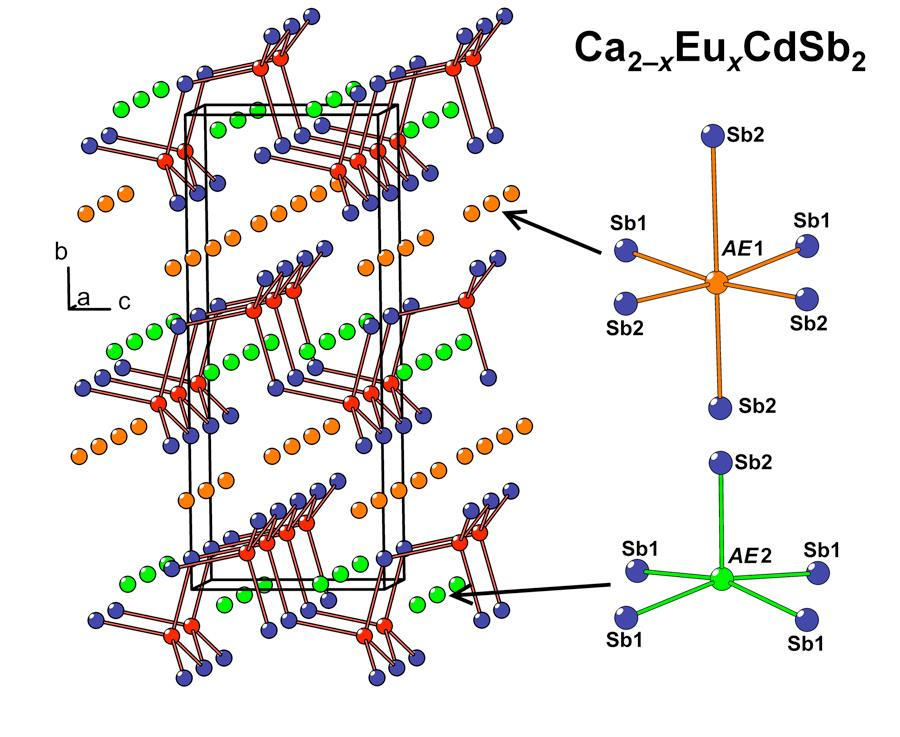
In the crystal structure of Ca
2−xEu
xCdSb
2 (
x ≈ 0.6), two nonequivalent
AE sites (
AE = Ca + Eu) are located between the Cd-Sb layers (
AE1) and within the layers (
AE2), respectively. The former site adopts a distorted octahedral coordination by the Sb atoms, while the latter position displays a square-pyramidal coordination (
Figure 2b). Due to the substantially different volumes of these two coordination polyhedra, pronounced preferred occupation is observed. The
AE1 site, offering a larger available volume, accumulates most of the Eu in the structure, whereas the smaller
AE2 site shows a minute Eu occupancy of about 2%, in line with a larger ionic radius of Eu
2+ versus Ca
2+. A similar trend was previously observed for the Yb
2CdSb
2-type solid solutions, Sr
2−xAxCdSb
2 (
A = Ca, Yb), Ba
2−xAxCdSb
2 (
A = Ca, Sr, Eu, Yb) [
39], and Yb
2−xEu
xCdSb
2 [
24,
39].
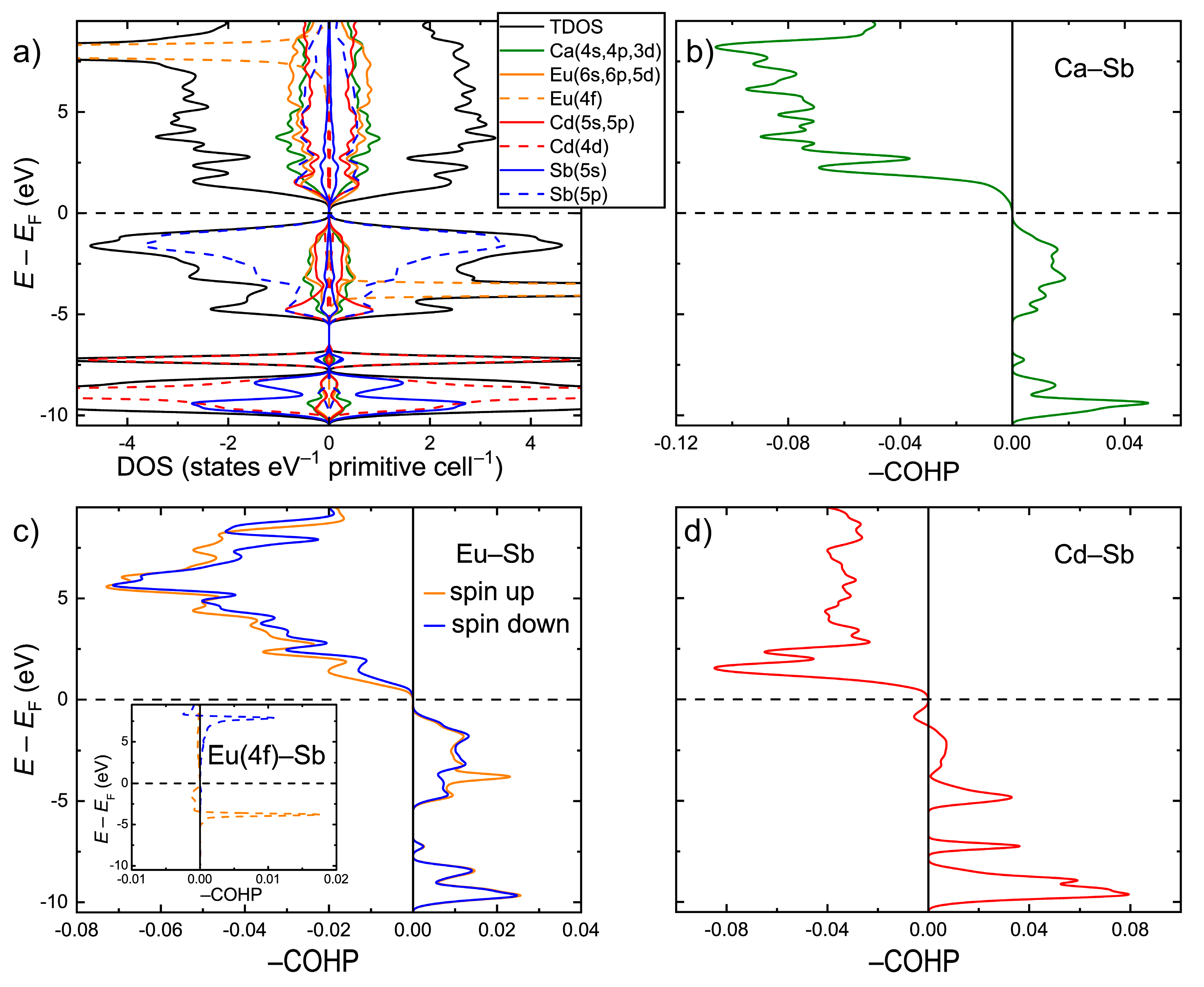
Electronic structure calculations were performed within the GGA+
U scheme for the idealized CaEuCdSb
2 composition after optimizing the lattice parameters and atomic coordinates. The electronic density of states (DOS) is shown in
Figure 3a. In accordance with the simple electron counting in the frame of the Zintl concept, CaEuCdSb
2 displays an electronic bandgap of about 0.5 eV. This value is larger than the 0.1 eV bandgap calculated for the isostructural YbEuCdSb
2 [
24]. This is likely is an electronic effect due to electronegativity differences between Yb and Ca, suggesting lower iconicity of the bonding in the latter compound. The states just below the Fermi level (
EF) mainly show the Sb(5p) character, indicating a significant electron transfer onto the Sb atoms. A strong hybridization of these states with Cd(5s) and Cd(5p) occurs in the energy window, −5.5 eV <
E–
EF < 0. The s, p, and d states of Ca and Eu also demonstrate a sizeable contribution in this energy region. A high peak of the Eu(4f) character is localized around
E–
EF = −3.7 eV. The converged magnetic moment on the Eu atom is 7.0 µ
B, in accordance with the 4f
7 ground term of the Eu
2+ ion. Similar to the case of YbEuCdSb
2, the Eu f orbitals cannot be considered completely inert in this case, since an apparent hybridization with the Sb(5p) states takes place around the peak position. In addition, a “tail” of the 4f peak extends up to the top of the valence band, hybridizing further with the Sb states. Well below the Fermi level, the DOS is dominated by the Sb(5s) states, corresponding to the Sb lone pairs, and by the occupied Cd(4d) states.
Analysis of the chemical bonding with the aid of the Crystal Orbital Hamilton Population curves (COHP) revealed optimized bonding interactions for the Ca–Sb and Eu–Sb pairs (
Figure 3c,d). Most importantly, orbital-resolved COHP analysis showed a small, yet non-negligible contribution of the Eu(4f)–Sb orbital mixing to the overall Eu–Sb bonding. The hybridization of the Eu(4f) and Sb(5p) states around the center of the Eu(4f) peak results in a localized set of bonding states, whereas the hybridization just under
EF gives rise to a broad domain of antibonding character. This finding suggests that the typically core-level 4f orbitals may play a role in the crystal structure formation/selection. The involvement of the 4f states in chemical bonding can be important, alongside atomic size factors, for understanding the stability of compounds that are known to form only with certain rare-earth elements, such as the binary
RE3Bi
7 phases with
RE = Nd and Sm [
44]. The Cd–Sb contacts display predominantly bonding interactions below
EF, with a small domain of antibonding states in the near vicinity of the Fermi level (
Figure 3d). A similar bonding pattern was observed for the Cd–Sb interactions in the prototypic Yb
2CdSb
2 structure [
38].
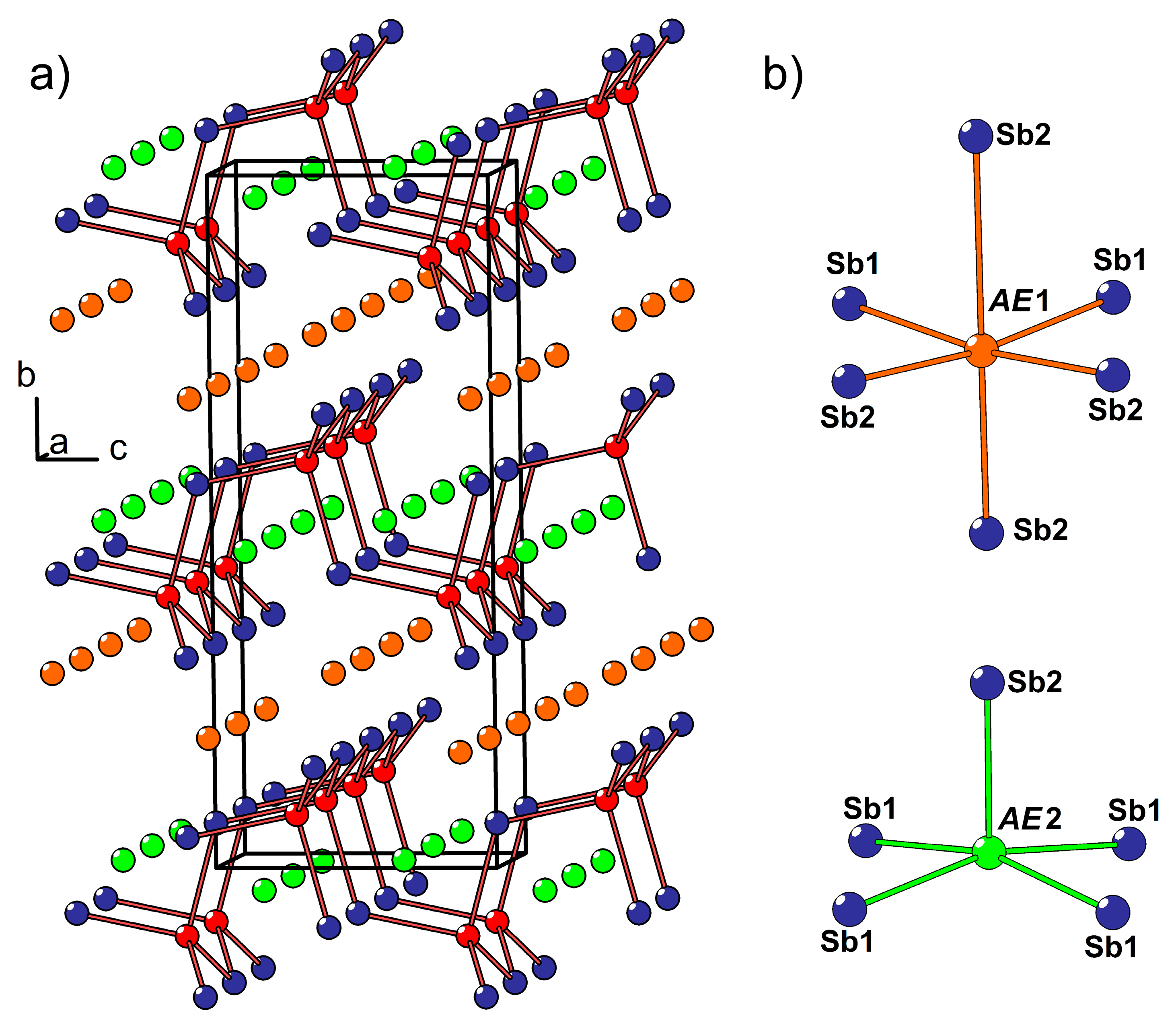
3.3. Crystal Structure of the Eu11−xCaxCd6Sb12 Solid Solution (x ≈ 1)
Eu
11−xCa
xCd
6Sb
12 crystallizes isotypically to the parent Eu
11Cd
6Sb
12 compound (Sr
11Cd
6Sb
12 type, Pearson code
mS58, space group
C2/
m (No. 12),
Figure 4a) [
45,
46]. The refined
x ≈ 1 in the studied crystal must be close to the upper limit of the Ca substitution for Eu, since the starting mixture contained a large surplus of Ca (recall that the nominal Eu: Ca ratio was 1:1). The crystal structure likely becomes unstable at higher Ca contents due to geometric/size factors. Accordingly, the pure ternary Ca phase has not been reported. This result is in line with the study on the isostructural solid solution, Eu
11−xYb
xCd
6Sb
12, where the Sr
11Cd
6Sb
12 type is retained only up to
x ≈ 2 [
47]. The similarity between the two quaternary systems is likely related to the close ionic radii of Ca
2+ and Yb
2+:
r(Ca
2+) = 1.00 Å,
r(Yb
2+) = 1.02 Å for coordination number 6 [
37].
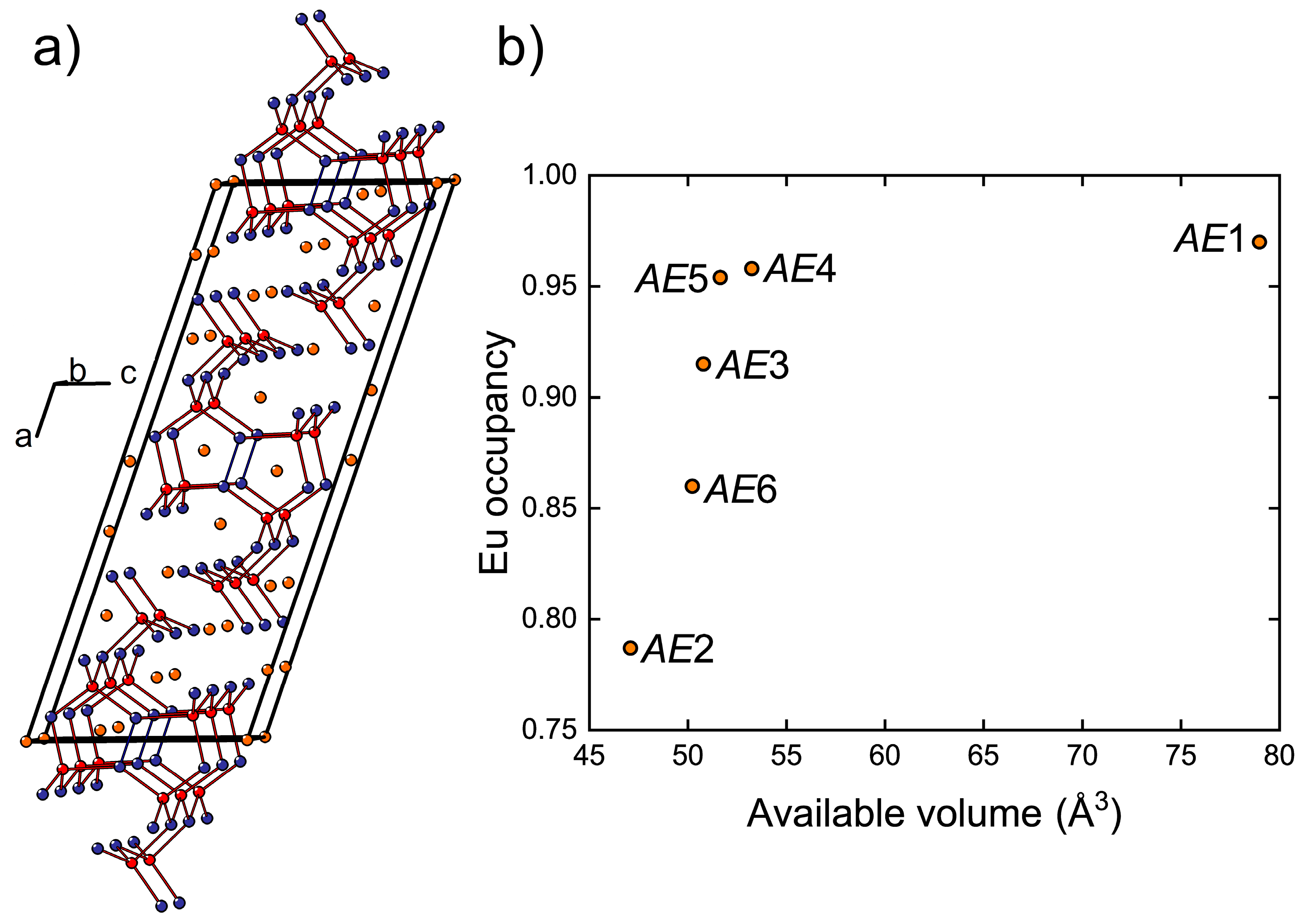
The crystal structure of Eu
11Cd
6Sb
12 and isostructural compositions has been described in detail previously [
45,
46,
47,
48,
49]. The anionic substructure contains infinite fragments of corner sharing CdSb
4 tetrahedra propagating along the monoclinic
b axis (
Figure 4a). Homoatomic Sb–Sb bonding develops within these fragments, resulting in Sb
2 dumbbells shared between adjacent Cd atoms. In Eu
11−xCa
xCd
6Sb
12, the
AE atoms (
AE = Eu + Ca), except
AE5, are six-fold coordinated by Sb.
AE5 resides in a five-fold coordination environment with an average
AE–Sb bond length of 3.35 Å. A six-fold coordination of
AE5 can be completed by including an Sb atom located at a much longer distance of 3.98 Å. Similarly, the six-fold coordination of the
AE1 site (<
d(
AE–Sb)> = 3.43 Å) can be regarded as a highly distorted square antiprism (coordination number 8) with two additional Sb atoms at 4.02 Å and 4.03 Å from
AE1. Although such long distances can hardly be considered bonding interactions, when considered, they provide an estimate of the available volume offered by the Sb coordination environment around each
AE position. In fact, the occupancy of the
AE sites by Eu correlates well with the volume of such “extended” polyhedra (
Figure 4b). The largest
AE1 site is occupied mainly by Eu, whereas the highest Ca occupancy is found for the most spatially confined
AE2 position.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}