Protective Carbon Overlayers from 2,3-Naphthalenediol Pyrolysis on Mesoporous SiO2 and Al2O3 Analyzed by Solid-State NMR


Abstract
: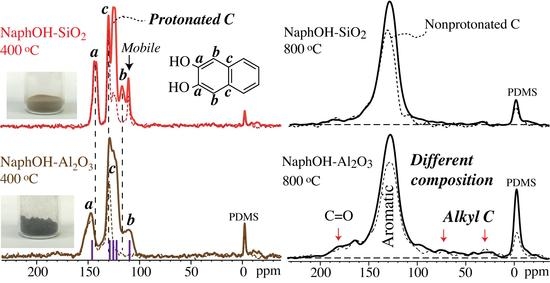
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Carbon Overlayers on Mesoporous Oxides
2.3. Tricalcium Phosphate Filler for NMR
2.4. Solid-State NMR Methods
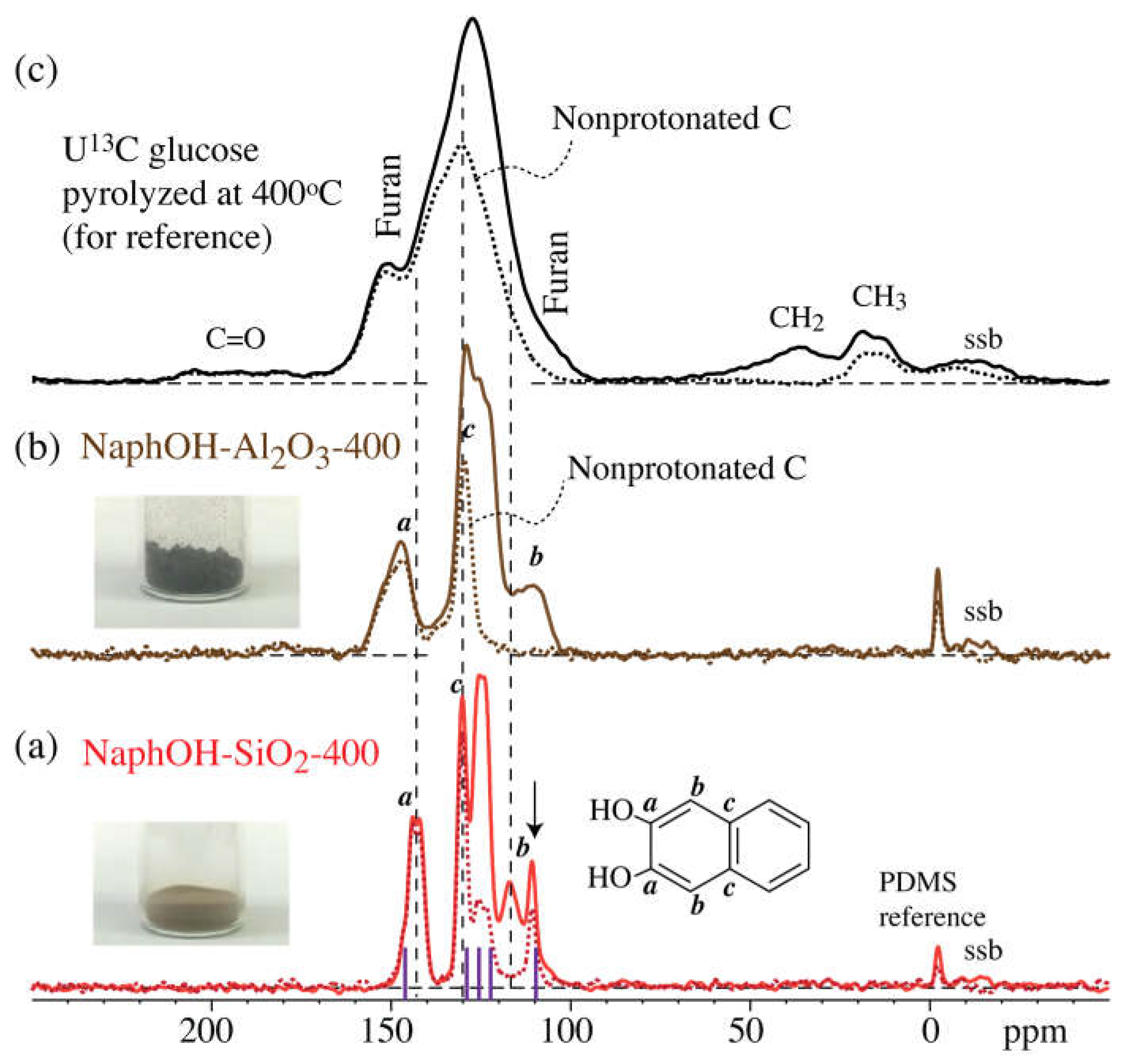
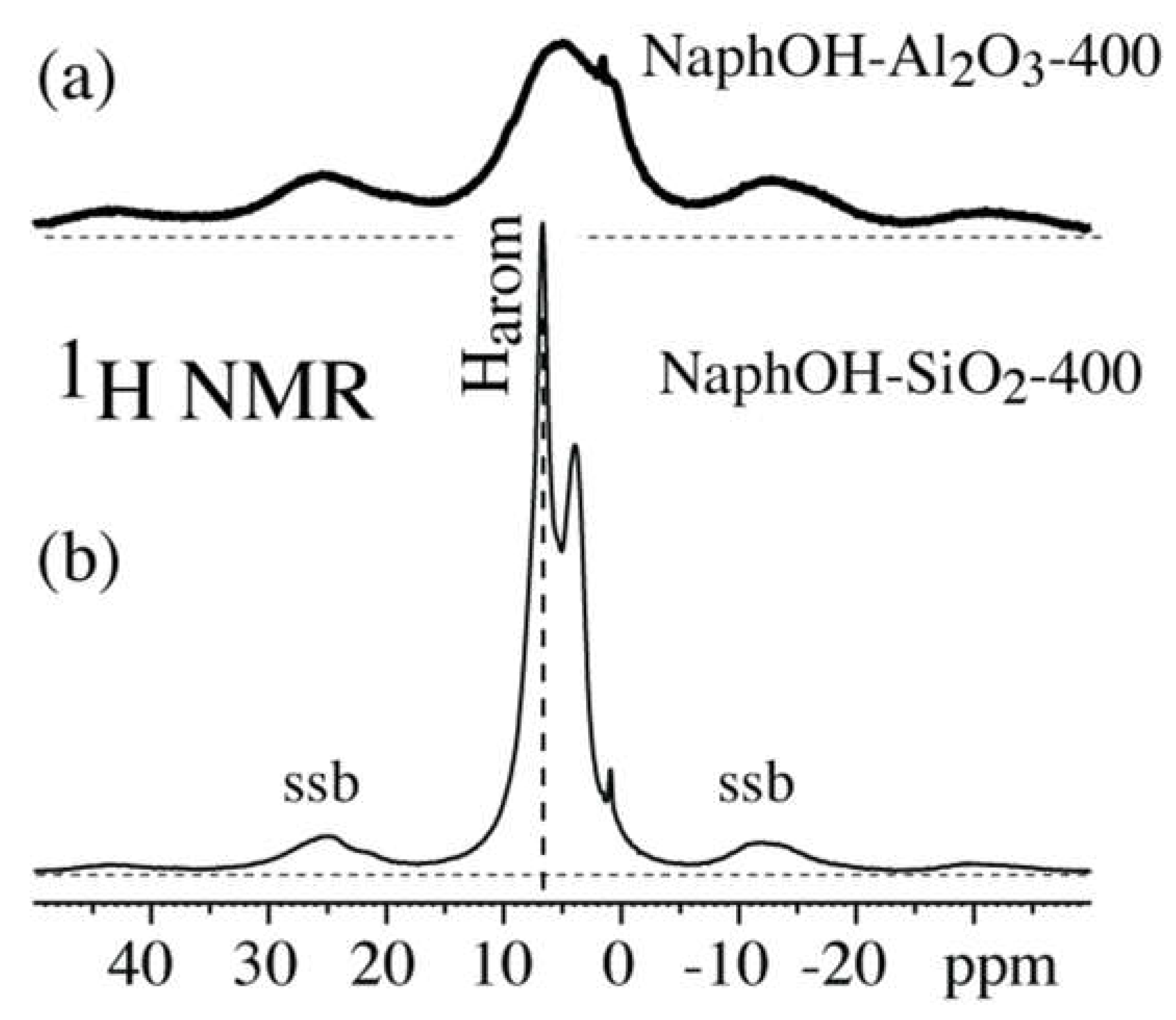
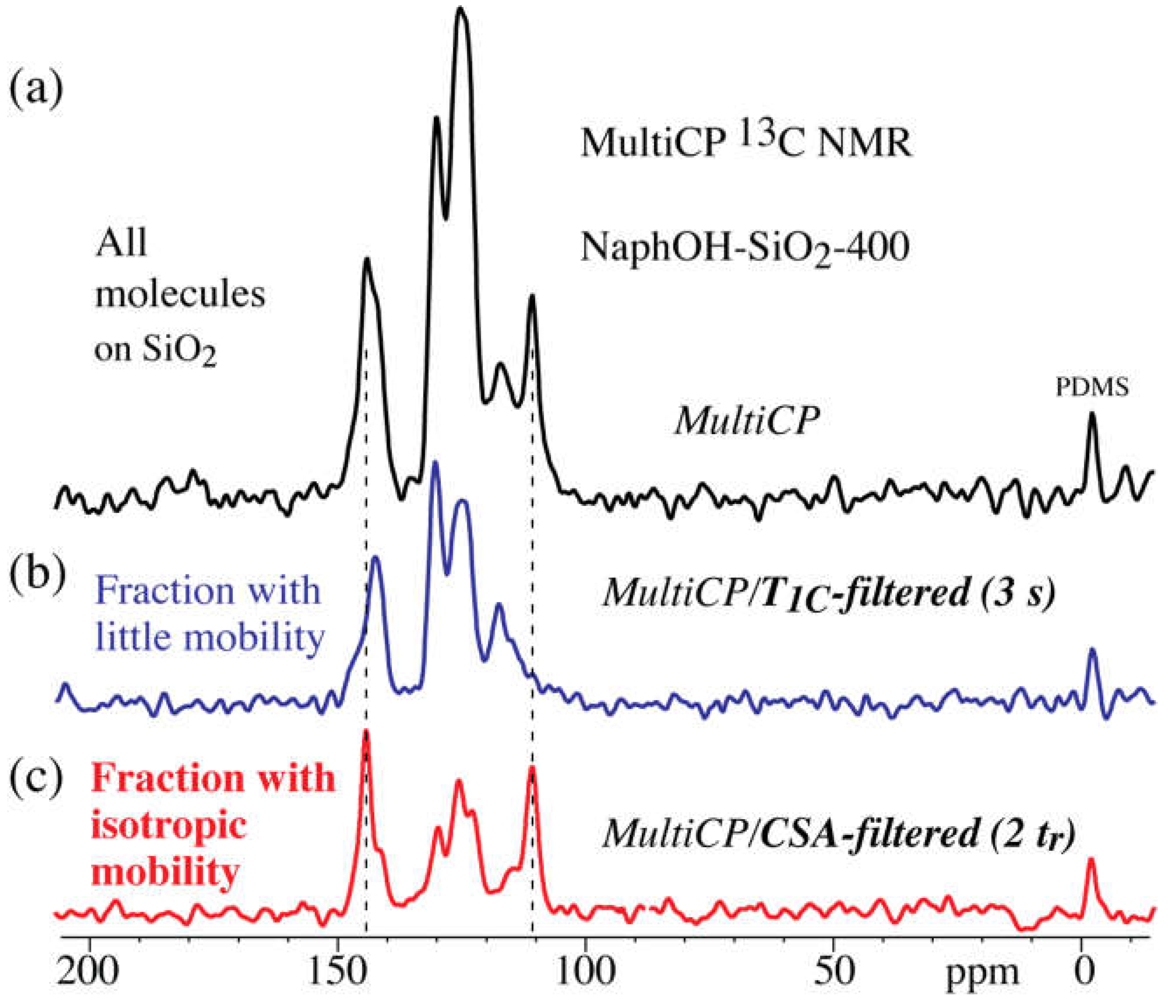
3. Results
4. Discussion
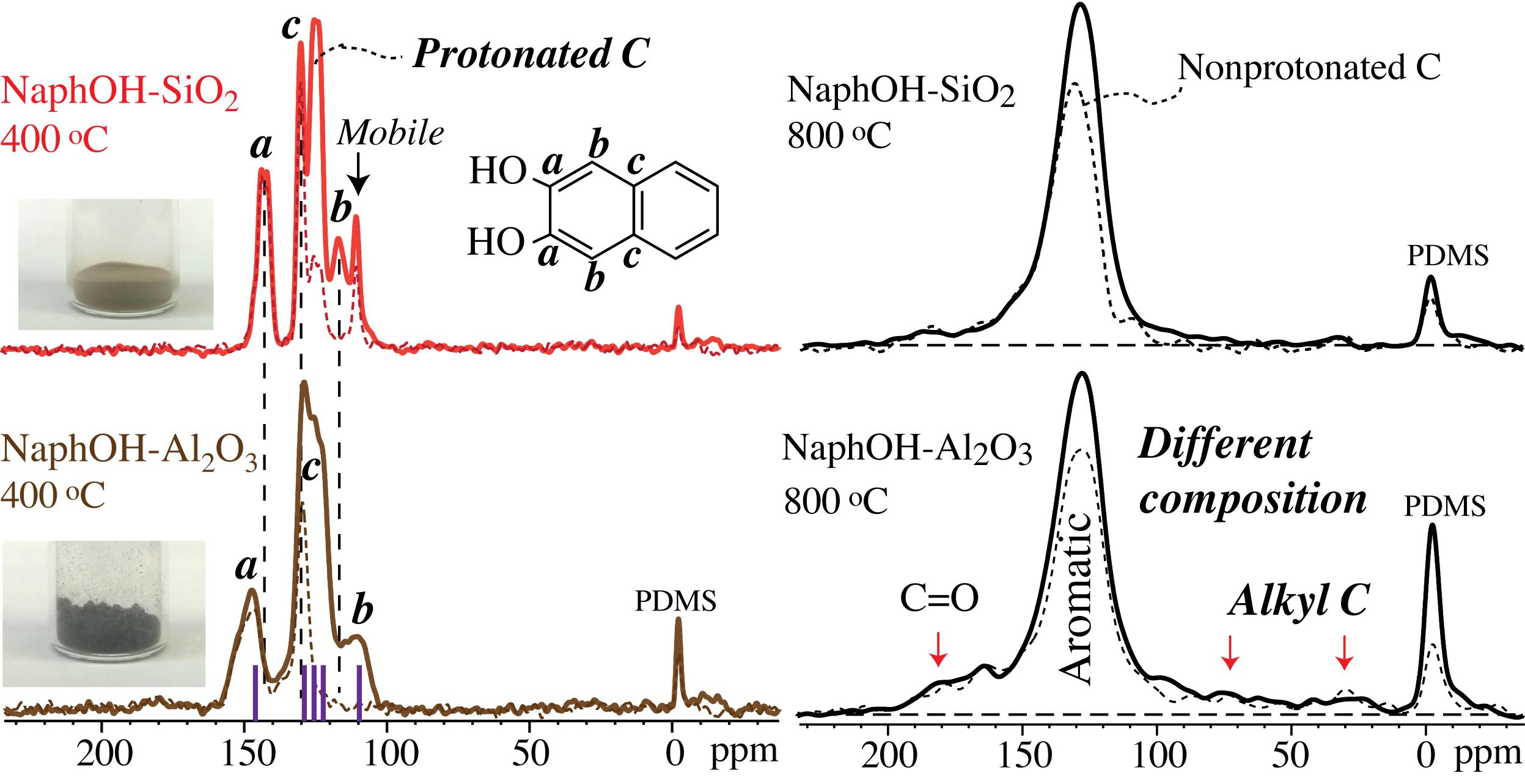
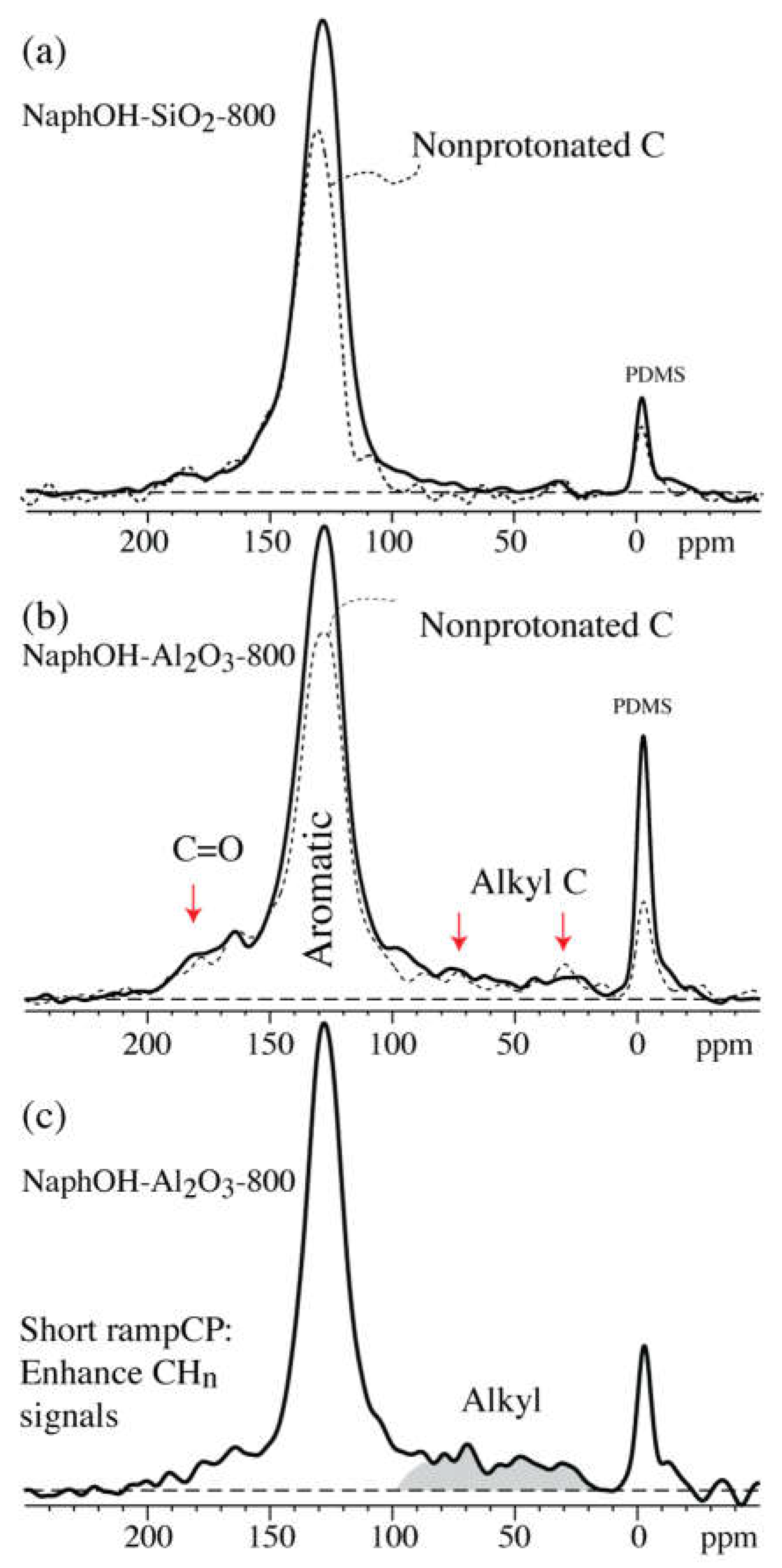
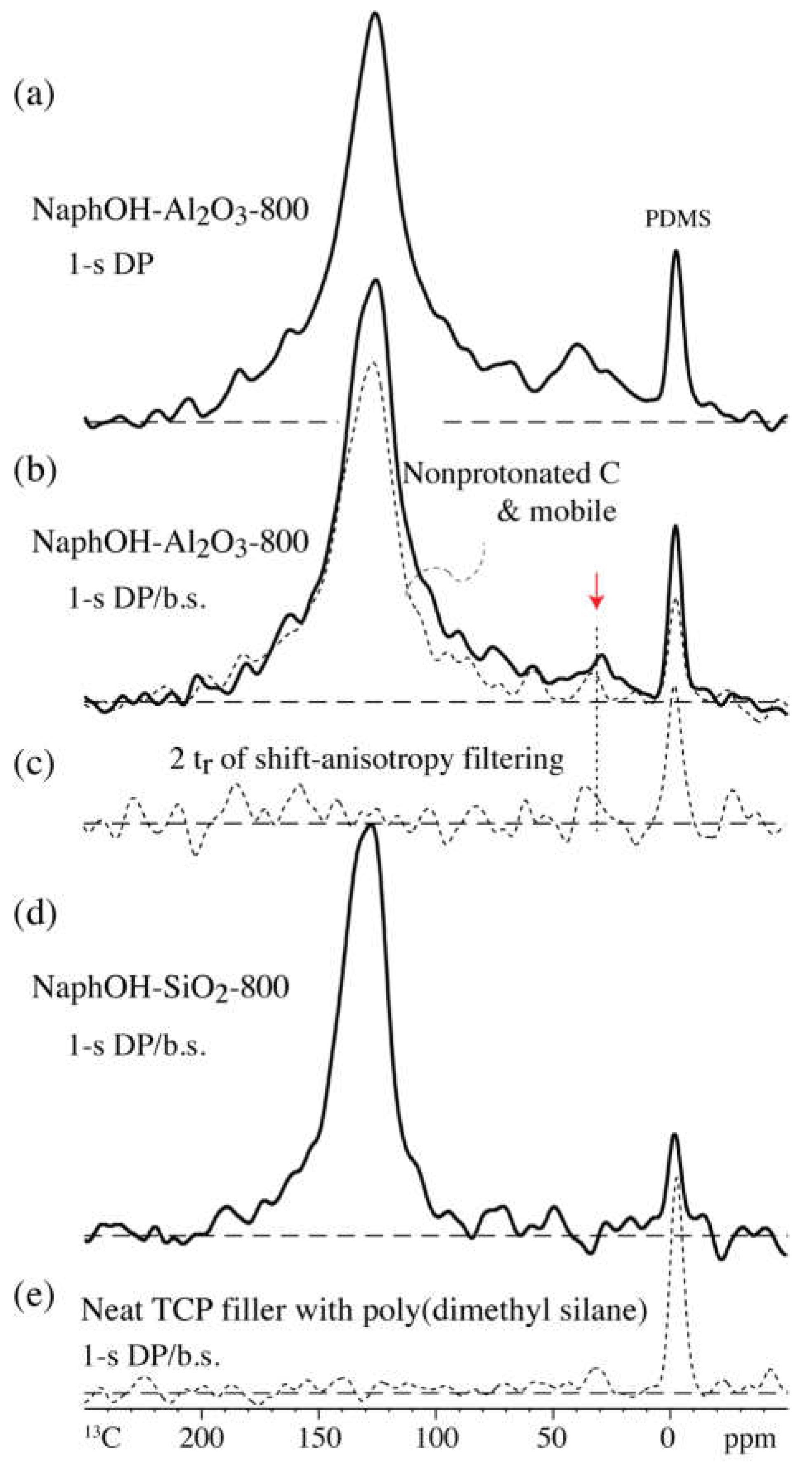
4.1. Comparison of Carbon Overlayers on Al2O3 vs. SiO2 at 800 °C
4.2. Tetrahedral Carbon Species Formed on the Al2O3 Support at 800 °C
4.3. Structure after 400 °C Heat Treatment
4.4. Highly Mobile Naphthalenediol on SiO2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098. [Google Scholar] [CrossRef] [PubMed]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Pham, H.N.; Datye, A.K. Hydrothermally stable heterogeneous catalysts for conversion of biorenewables. Green Chem. 2014, 16, 4627–4643. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of Biomass into Chemicals over Metal Catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Pagan-Torres, Y.J.; Gallo, J.M.R.; Wang, D.; Pham, H.N.; Libera, J.A.; Marshall, C.L.; Elam, J.W.; Datye, A.K.; Dumesic, J.A. Synthesis of Highly Ordered Hydrothermally Stable Mesoporous Niobia Catalysts by Atomic Layer Deposition. ACS Catal. 2011, 1, 1234–1245. [Google Scholar] [CrossRef]
- Pham, H.N.; Anderson, A.E.; Johnson, R.L.; Schmidt-Rohr, K.; Datye, A.K. Improved Hydrothermal Stability of Mesoporous Oxides for Reactions in the Aqueous Phase. Angew. Chem. Int. Ed. 2012, 51, 13163–13167. [Google Scholar] [CrossRef] [PubMed]
- Ravenelle, R.M.; Copeland, J.R.; Kim, W.G.; Crittenden, J.C.; Sievers, C. Structural Changes of γ-Al2O3-Supported Catalysts in Hot Liquid Water. ACS Catal. 2011, 1, 552–561. [Google Scholar] [CrossRef]
- Ravenelle, R.M.; Copeland, J.R.; Van Pelt, A.H.; Crittenden, J.C.; Sievers, C. Stability of Pt/γ-Al2O3 Catalysts in Model Biomass Solutions. Top. Catal. 2012, 55, 162–174. [Google Scholar] [CrossRef]
- Pham, H.N.; Anderson, A.E.; Johnson, R.L.; Schwartz, T.J.; O'Neill, B.J.; Duan, P.; Schmidt-Rohr, K.; Dumesic, J.A.; Datye, A.K. Carbon Overcoating of Supported Metal Catalysts for Improved Hydrothermal Stability. ACS Catal. 2015, 5, 4546–4555. [Google Scholar] [CrossRef]
- Anderson, J.M.; Johnson, R.L.; Schmidt-Rohr, K.; Shanks, B.H. Solid state NMR study of chemical structure and hydrothermal deactivation of moderate-temperature carbon materials with acidic SO3H sites. Carbon 2014, 74, 333–345. [Google Scholar] [CrossRef]
- Johnson, R.L.; Anderson, J.M.; Shanks, B.H.; Fang, X.; Hong, M.; Schmidt-Rohr, K. Spectrally edited 2D 13C-13C NMR spectra without diagonal ridge for characterizing 13C-enriched low-temperature carbon materials. J. Magn. Reson. 2013, 234, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.E.; Schmidt-Rohr, K.; Satrio, J.A.; Brown, R.C. Characterization of biochar from fast pyrolysis and gasification systems. Environ. Progr. Sustain. Energy 2009, 28, 386–396. [Google Scholar] [CrossRef]
- Baccile, N.; Falco, C.; Titirici, M.M. Characterization of biomass and its derived char using 13C-solid state nuclear magnetic resonance. Green Chem. 2014, 16, 4839–4869. [Google Scholar] [CrossRef]
- Mochalin, V.; Osswald, S.; Gogotsi, Y. Contribution of functional groups to the Raman spectrum of nanodiamond powder. Chem. Mater. 2009, 21, 273–279. [Google Scholar] [CrossRef]
- Chen, J.; Deng, S.Z.; Chen, J.; Yu, Z.X.; Xu, N.S. Graphitization of nanodiamond powder annealed in argon ambient. Appl. Phys. Lett. 1999, 74, 3651–3653. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K.; Cui, J.-F. Reply to "Comment on 'Quantification of C=C and C=O Surface Carbons in Detonation Nanodiamond by NMR'". J. Phys. Chem. C 2014, 119, 21288–21291. [Google Scholar] [CrossRef]
- Huo, J.; Johnson, R.L.; Duan, P.; Pham, H.N.; Mendivelso-Perez, D.; Smith, E.A.; Datye, A.K.; Schmidt-Rohr, K.; Shanks, B.H. Stability of Pd nanoparticles on carbon-coated supports under hydrothermal conditions. Catal. Sci. Technol. 2018, 8, 1151–1160. [Google Scholar] [CrossRef]
- Osswald, S.; Yushin, G.; Mochalin, V.; Kucheyev, S.O.; Gogotsi, Y. Control of sp2/sp3 Carbon Ratio and Surface Chemistry of Nanodiamond Powders by Selective Oxidation in Air. J. Am. Chem. Soc. 2006, 128, 11635–11642. [Google Scholar] [CrossRef] [PubMed]
- Newville, M. Fundamentals of XAFS; University of Chicago: Chicago, IL, USA, 2004. [Google Scholar]
- Soderlind, E.; Stilbs, P. A 2H NMR study of two cationic surfactants adsorbed on silica particles. Langmuir 1993, 9, 2024–2034. [Google Scholar] [CrossRef]
- Johnson, R.L.; Schwartz, T.J.; Dumesic, J.A.; Schmidt-Rohr, K. Methionine bound to Pd/γ-Al2O3 catalysts studied by solid-state 13C NMR. Solid State Nucl. Magn. Reson. 2015, 72, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Fukura, Y.; Inde, K.; Tsuji, K.; Takeuchi, M.; Kyotani, T. Carbon-coated mesoporous silica with hydrophobicity and electrical conductivity. Carbon 2008, 46, 48–53. [Google Scholar] [CrossRef]
- Nishihara, H.; Kwon, T.; Fukura, Y.; Nakayama, W.; Hoshikawa, Y.; Iwamura, S.; Nishiyama, N.; Itoh, T.; Kyotani, T. Fabrication of a Highly Conductive Ordered Porous Electrode by Carbon-Coating of a Continuous Mesoporous Silica Film. Chem. Mater. 2011, 23, 3144–3151. [Google Scholar] [CrossRef]
- Duan, P.; Schmidt-Rohr, K. Composite-pulse and partially dipolar dephased multiCP for improved quantitative solid-state 13C NMR. J. Magn. Reson. 2017, 285, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.D.; Schmidt-Rohr, K. Accurate Quantification of Aromaticity and Nonprotonated Aromatic Carbon Fraction in Natural Organic Matter by 13C Solid-State Nuclear Magnetic Resonance. Environ. Sci. Technol. 2004, 38, 2680–2684. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.-D.; Schmidt-Rohr, K. Separation of aromatic-carbon 13C NMR signals from di-oxygenated alkyl bands by a chemical-shift-anisotropy filter. Solid State Nucl. Magn. Reson. 2004, 26, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Bodenhausen, G.; Freeman, R.; Turner, D.L. Suppression of artifacts in two-dimensional J spectroscopy. J. Magn. Reson. 1977, 27, 511–514. [Google Scholar] [CrossRef]
- Hahn, E.L. Spin Echoes. Phys. Rev. 1950, 80, 580–594. [Google Scholar] [CrossRef]
- Chen, Q.; Hou, S.S.; Schmidt-Rohr, K. A simple scheme for probehead background suppression in one-pulse 1H NMR. Solid State Nucl. Magn. Reson. 2004, 26, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Abragam, A. Principles of Nuclear Magnetism; Oxford University Press: Oxford, UK, 1961; pp. 171–193. [Google Scholar]
- Nayeem, A.; Yesinowski, J.P. Calculation of magic-angle spinning nuclear magnetic resonance spectra of paramagnetic solids. J. Chem. Phys. 1988, 89, 4600–4608. [Google Scholar] [CrossRef]
- Pan, H.; Pruski, M.; Gerstein, B.; Li, F.; Lannin, J.S. Local coordination of carbon atoms in amorphous carbon. Phys. Rev. B 1991, 44, 6741. [Google Scholar] [CrossRef]
- Brough, A.R.; Grey, C.P.; Dobson, C.M. Paramagnetic ions as structural probes in solid-state NMR: distance measurements in crystalline lanthanide acetates. J. Am. Chem. Soc. 1993, 115, 7318–7327. [Google Scholar] [CrossRef]
- Gwendal, K.; Anthony, D.A.; Loïc, T.; Olivier, M.; Lyndon, E.; Guido, P. Crystal-Structure Determination of Powdered Paramagnetic Lanthanide Complexes by Proton NMR Spectroscopy. Angew. Chem. Int. Ed. 2009, 48, 3082–3086. [Google Scholar] [CrossRef]
- Walder, B.J.; Dey, K.K.; Davis, M.C.; Baltisberger, J.H.; Grandinetti, P.J. Two-dimensional NMR measurement and point dipole model prediction of paramagnetic shift tensors in solids. J. Chem. Phys. 2015, 142, 014201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnet, C.; Fontaine, J.; Lefèbvre, F.; Grill, A.; Patel, V.; Jahnes, C. Solid state 13C and 1H nuclear magnetic resonance investigations of hydrogenated amorphous carbon. J. Appl. Phys. 1999, 85, 3264–3270. [Google Scholar] [CrossRef]
- Peng, J.; Sergiienko, A.; Mangolini, F.; Stallworth, P.E.; Greenbaum, S.; Carpick, R.W. Solid state magnetic resonance investigation of the thermally-induced structural evolution of silicon oxide-doped hydrogenated amorphous carbon. Carbon 2016, 105, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Hoffmann, R.; Ashcroft, N.W.; Badding, J.; Xu, E.; Crespi, V. Linearly Polymerized Benzene Arrays As Intermediates, Tracing Pathways to Carbon Nanothreads. J. Am. Chem. Soc. 2015, 137, 14373–14386. [Google Scholar] [CrossRef] [PubMed]
- Donnet, J.-B.; Fousson, E.; Delmotte, L.; Samirant, M.; Baras, C.; Wang, T.K.; Eckhardt, A. 13C NMR characterization of nanodiamonds. C.R. Acad. Sci., Ser. IIc: Chim. 2000, 3, 831–838. [Google Scholar] [CrossRef]
- Fang, X.; Mao, J.; Levin, E.M.; Schmidt-Rohr, K. Nonaromatic Core-Shell Structure of Nanodiamond from Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.M.; Paul, E.G. Carbon-13 Magnetic Resonance. II. Chemical Shift Data for the Alkanes. J. Am. Chem. Soc. 1964, 86, 2984–2990. [Google Scholar] [CrossRef]
- Bovey, F.A.; Mirau, P.A. NMR of Polymers; Academic Press: San Diego, CA, USA, 1996; p. 459. [Google Scholar]
- Golzan, M.; Lukins, P.; McKenzie, D.; Vassallo, A.; Hanna, J. NMR evidence for strained carbon bonding in tetrahedral amorphous carbon. Chem. Phys. 1995, 193, 167–172. [Google Scholar] [CrossRef]
- Alam, T.M.; Friedmann, T.A.; Jurewicz, A.J. Solid State 13C MAS NMR Investigations of Amorphous Carbon Thin Films. In Thin Films: Preparation, Characterization, Applications; Soriaga, M.P., Stickney, J., Bottomley, L.A., Kim, Y.-G., Eds.; Springer Science & Business Media: New York, NY, USA, 2002; pp. 277–289. [Google Scholar]
- Alam, T.M.; Friedmann, T.; Schultz, P.A.; Sebastiani, D. Low temperature annealing in tetrahedral amorphous carbon thin films observed by 13C NMR spectroscopy. Phys. Rev. B 2003, 67, 245309. [Google Scholar] [CrossRef]
- Alam, T.M. Solid-state 13C magic angle spinning NMR spectroscopy characterization of particle size structural variations in synthetic nanodiamonds. Mater. Chem. Phys. 2004, 85, 310–315. [Google Scholar] [CrossRef]
- Shames, A.I.; Panich, A.M.; Kempinski, W.; Alexenskii, A.E.; Baidakova, M.V.; Dideikin, A.T.; Osipov, V.Y.; Siklitski, V.I.; Osawa, E.; Ozawa, M.; Vul, A.Y. Defects and impurities in nanodiamonds: EPR, NMR and TEM study. J. Phys. Chem. Solids 2002, 63, 1993–2001. [Google Scholar] [CrossRef]
- Levin, E.M.; Fang, X.W.; Bud'ko, S.L.; Straszheim, W. E.; McCallum, R.W.; Schmidt-Rohr, K. Magnetization and 13C NMR spin-lattice relaxation of nano-diamond powder. Physical Review B: Condensed Matter and Materials Physics 2008, 77, 054418/054410–054418/054411. [Google Scholar] [CrossRef]
- Dubois, M.; Guerin, K.; Batisse, N.; Petit, E.; Hamwi, A.; Komatsu, N.; Kharbache, H.; Pirotte, P.; Masin, F. Solid State NMR study of nanodiamond surface chemistry. Solid State Nucl. Magn. Reson. 2011, 40, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Panich, A.M.; Shames, A.I.; Vieth, H.M.; Osawa, E.; Takahashi, M.; Vul, A.Y. Nuclear magnetic resonance study of ultrananocrystalline diamonds. Eur. Phys. J. B 2006, 52, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Panich, A.M.; Furman, G.B. Nuclear spin–lattice relaxation and paramagnetic defects in carbon nanomaterials. Diamond Relat. Mater. 2012, 23, 157–161. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, P.; Cao, X.; Pham, H.; Datye, A.; Schmidt-Rohr, K. Protective Carbon Overlayers from 2,3-Naphthalenediol Pyrolysis on Mesoporous SiO2 and Al2O3 Analyzed by Solid-State NMR. Materials 2018, 11, 980. https://doi.org/10.3390/ma11060980
Duan P, Cao X, Pham H, Datye A, Schmidt-Rohr K. Protective Carbon Overlayers from 2,3-Naphthalenediol Pyrolysis on Mesoporous SiO2 and Al2O3 Analyzed by Solid-State NMR. Materials. 2018; 11(6):980. https://doi.org/10.3390/ma11060980
Chicago/Turabian StyleDuan, Pu, Xiaoyan Cao, Hien Pham, Abhaya Datye, and Klaus Schmidt-Rohr. 2018. "Protective Carbon Overlayers from 2,3-Naphthalenediol Pyrolysis on Mesoporous SiO2 and Al2O3 Analyzed by Solid-State NMR" Materials 11, no. 6: 980. https://doi.org/10.3390/ma11060980
APA StyleDuan, P., Cao, X., Pham, H., Datye, A., & Schmidt-Rohr, K. (2018). Protective Carbon Overlayers from 2,3-Naphthalenediol Pyrolysis on Mesoporous SiO2 and Al2O3 Analyzed by Solid-State NMR. Materials, 11(6), 980. https://doi.org/10.3390/ma11060980