Sintering Temperature-Dependence on Radiopacity of Bi(2−x) ZrxO(3+x/2) Powders Prepared by Sol-Gel Process

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
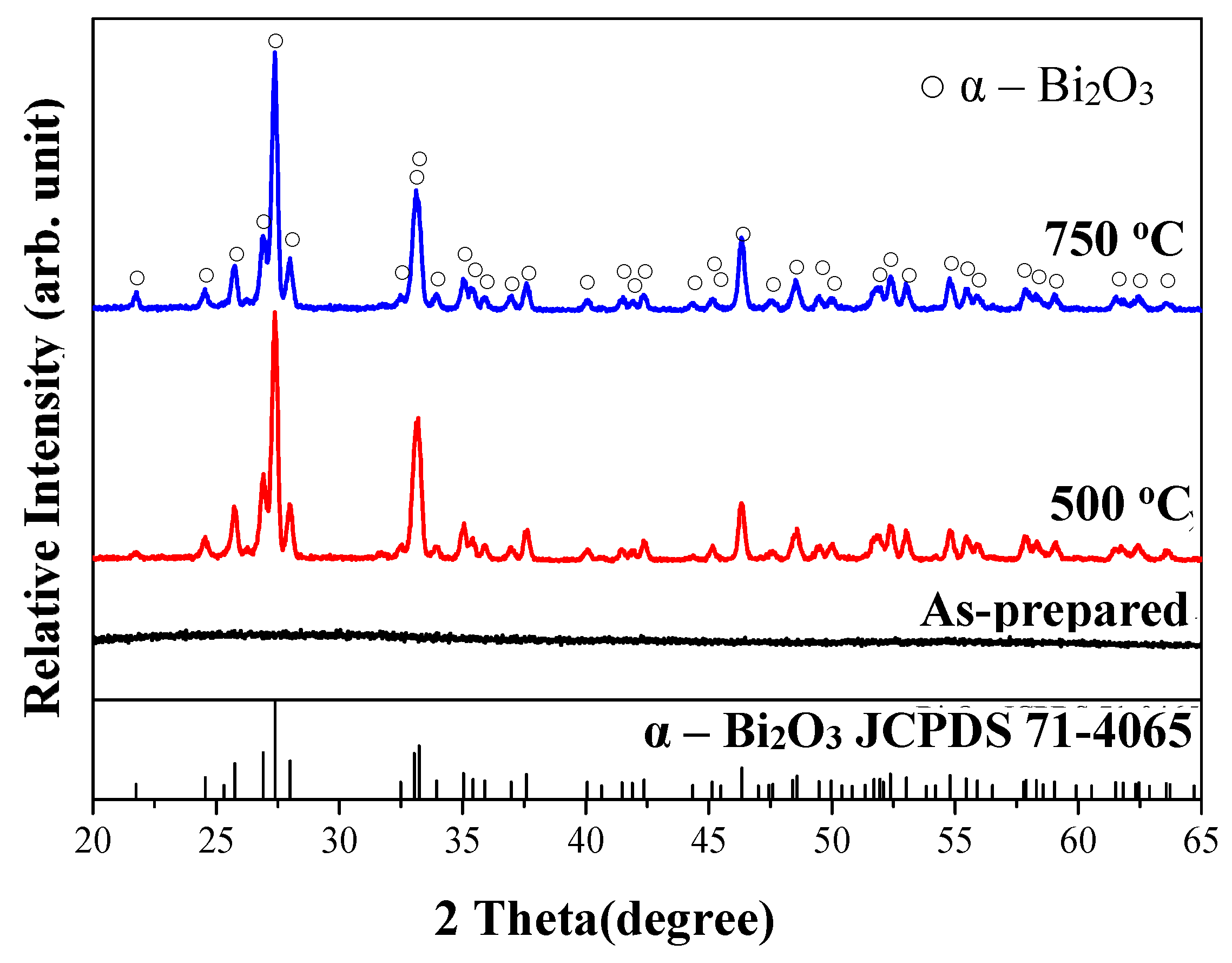
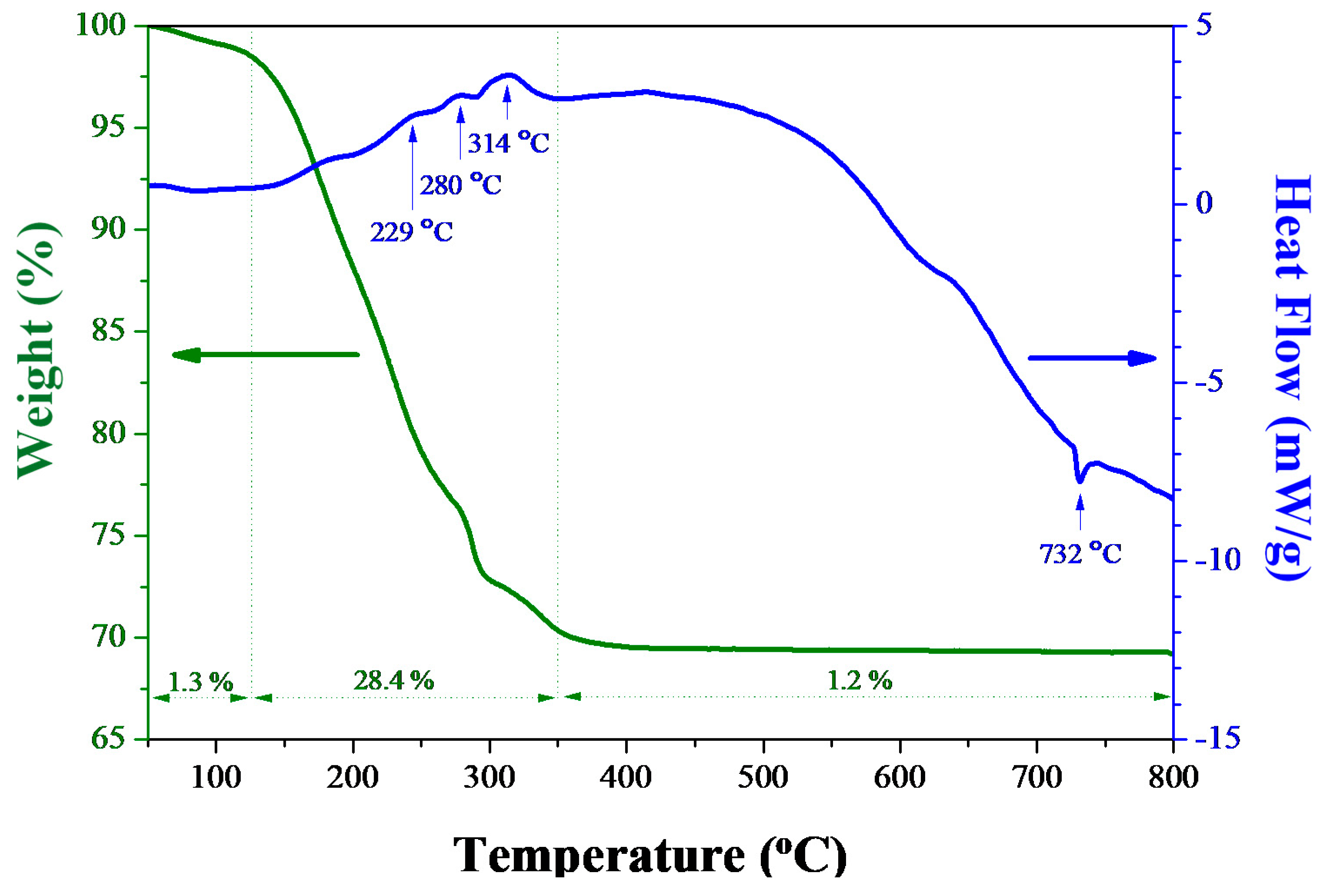
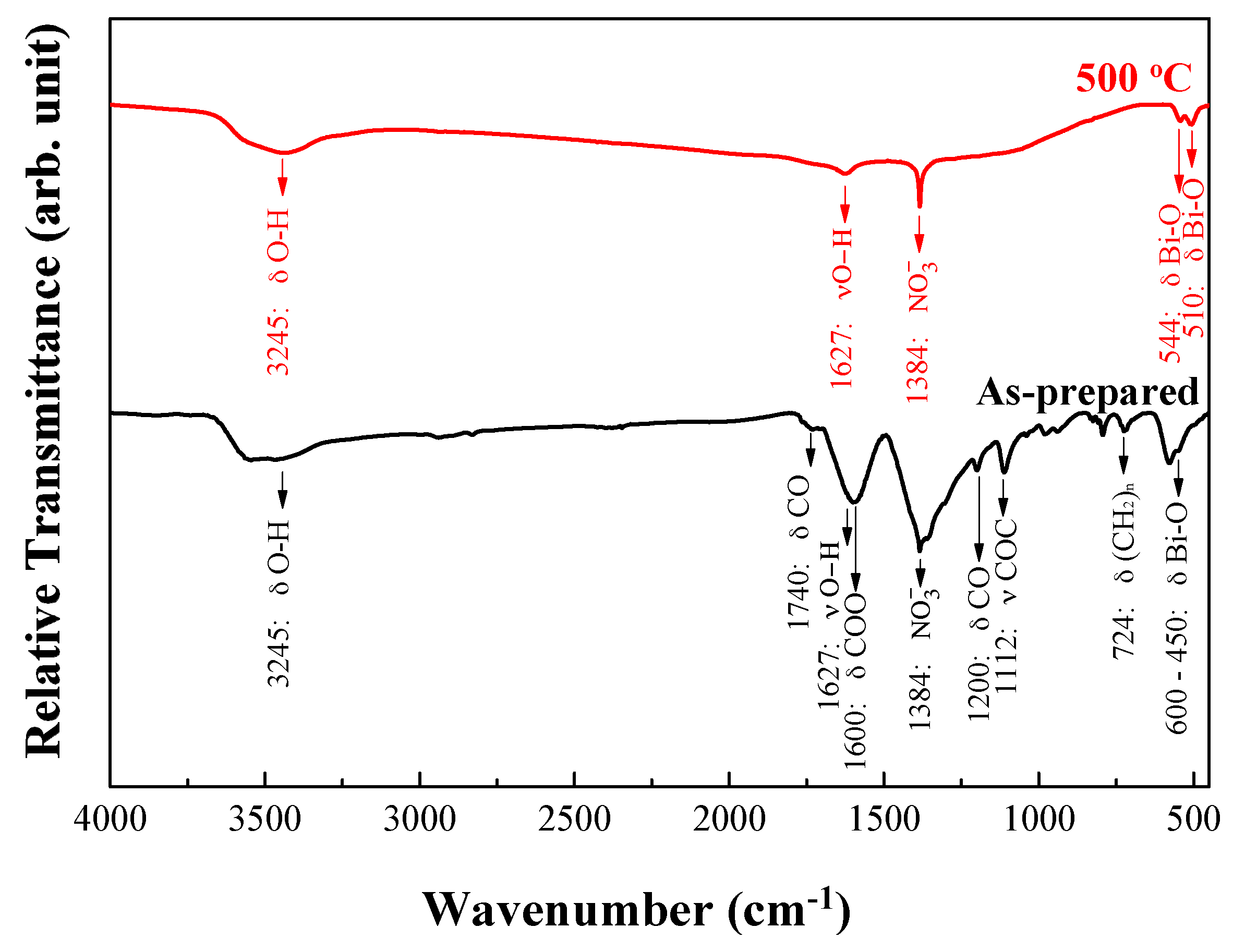
3.1. Characterization of As-Fabricated Bi2O3 Powders
3.2. Thermal Properties
3.3. Crystallization and Microstructure
3.4. FT-IR Analyses
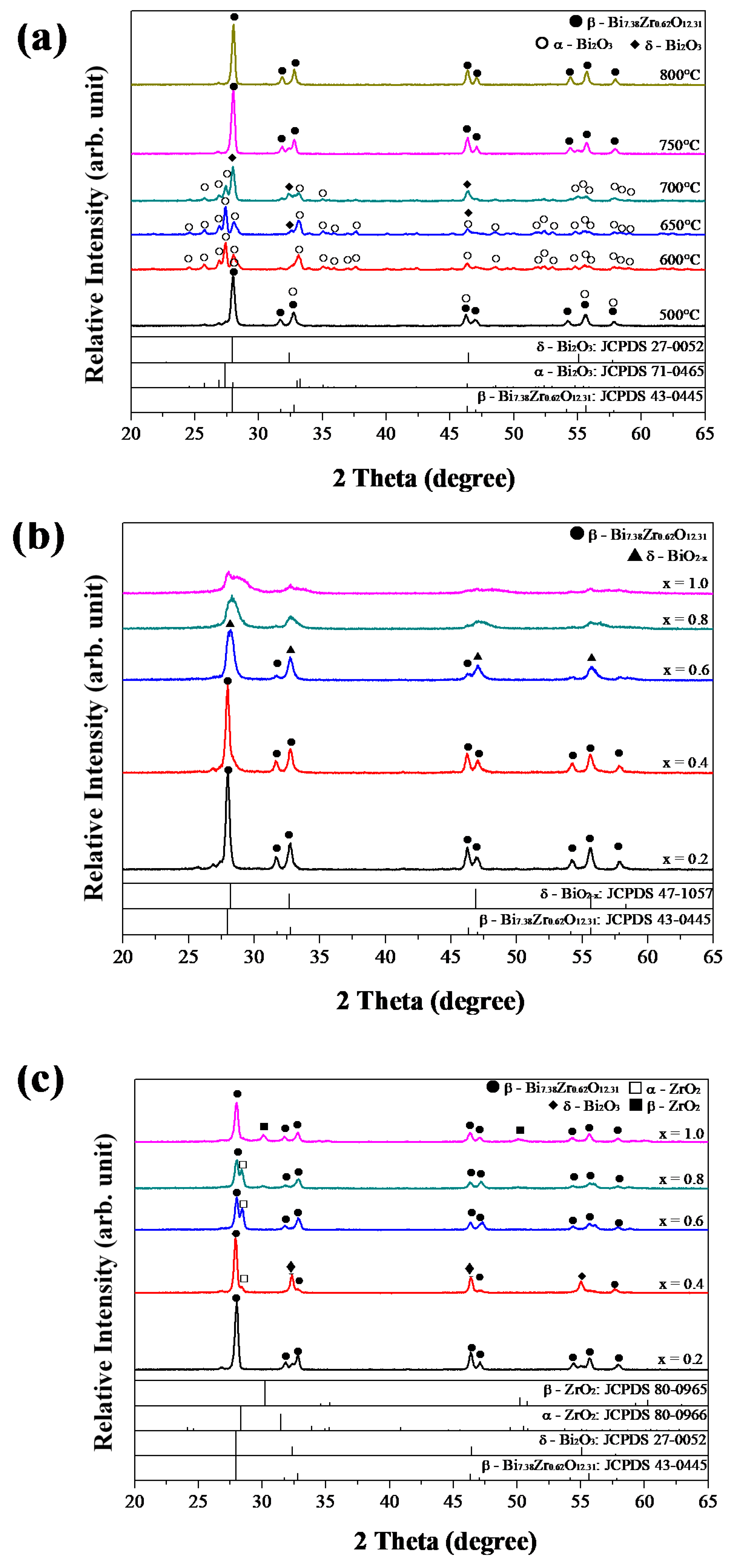
3.5. Characterization of Bi2−xZrxO3+x/2 Powders
3.6. XRD Analyses
3.7. Morphology and Microstructure
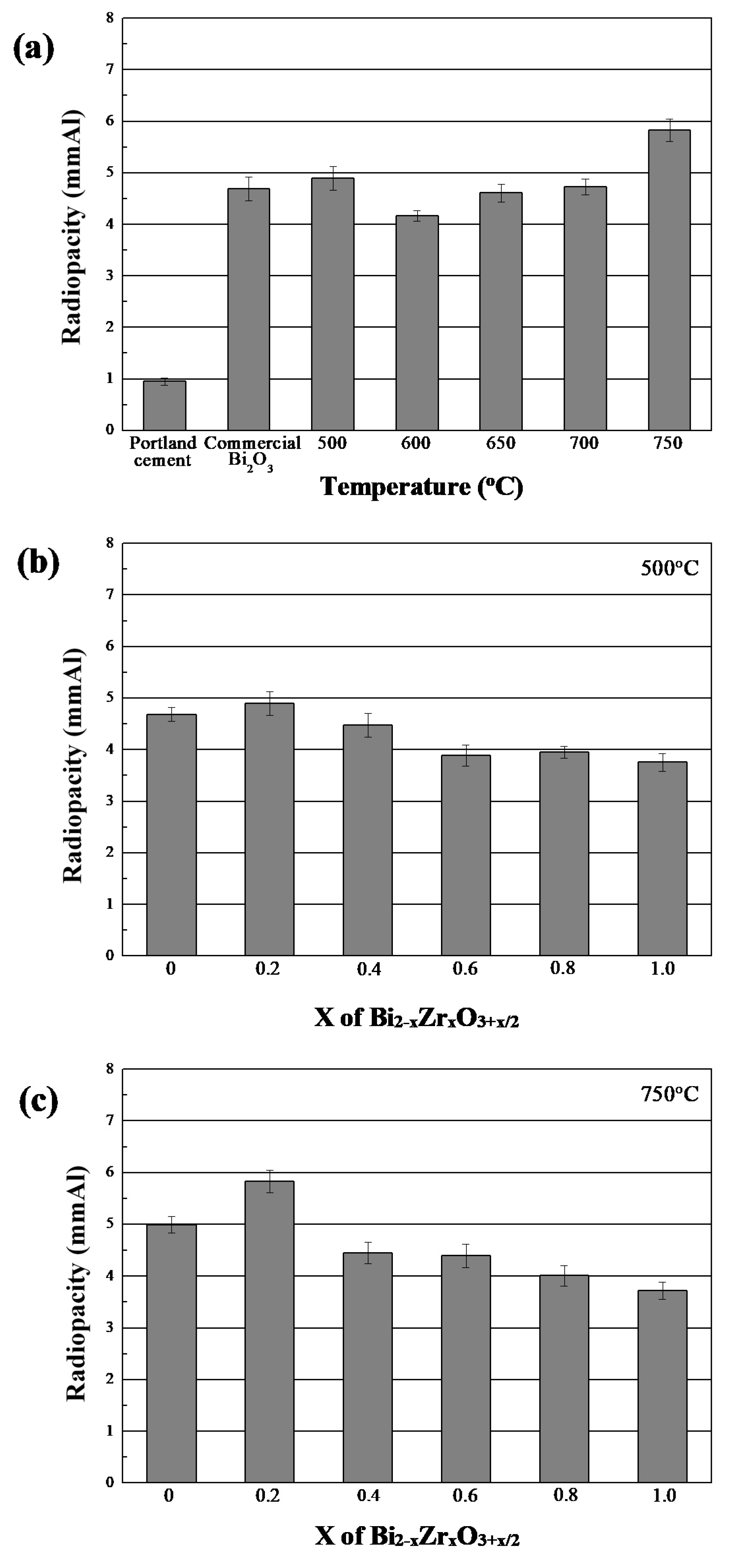
3.8. Radiopacity of Bi2−xZrxO3+x/2 Powders
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pekkan, G. Radiopacity of Dental Materials: An Overview. Avicenna J. Dent. Res. 2016, 8, e36847. [Google Scholar] [CrossRef]
- Gu, S.; Rasimick, B.J.; Deutsch, A.S.; Musikant, B.L. Radiopacity of dental materials using a digital X-ray system. Dent. Mater. 2006, 22, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Torabinejad, M.; Hong, C.U.; McDonald, F.; Pitt Ford, T.R. Physical and chemical properties of a new root-end filling material. J. Endod. 1995, 21, 349–533. [Google Scholar] [CrossRef]
- Skartveit, L.; Halse, A. Radiopacity of glass ionomer materials. J. Oral Rehabil. 1996, 23, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Pires de Souza, F.C.; Pardini, L.C.; Cruvinel, D.R.; Hamida, H.M.; Garcia, L.F. In vitro comparison of the radiopacity of cavity lining materials with human dental structures. J. Conser. Dent. 2010, 13, 65–70. [Google Scholar] [CrossRef]
- Reis, J.M.; Jorge, E.G.; Ribeiro, J.G.; Pinelli, L.A.; Abi-Rached Fde, O.; Tanomaru-Filho, M. Radiopacity evaluation of contemporary luting cements by digitization of images. ISRN Dent. 2012, 704246. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, S.T.; Haasken, B. Radiopacity of impression materials. Oral Surg. Oral Med. Oral Pathol. 1979, 47, 485–491. [Google Scholar] [CrossRef]
- Van der Linden, L.W.; van Aken, J. The origin of localized increased radiopacity in the dentin. Oral Surg. Oral Med. Oral Pathol. 1973, 35, 862–871. [Google Scholar] [CrossRef]
- Powell, L.V. Composite-resin materials and techniques in dentistry. Curr. Opin. Dent. 1992, 2, 128–136. [Google Scholar] [PubMed]
- Dentistry—Polymer-Based Restorative Materials. ISO International Organization for Standardization 2009; ISO 4049; ISO: Geneva, Switzerland, 2009.
- Dental Root Canal Sealing Materials. ISO International Organization for Standardization 2012; ISO 6876; ISO: Geneva, Switzerland, 2012.
- Beyer-Olsen, E.M.; Orstavik, D. Radiopacity of root canal sealers. Oral Surg. Oral Med. Oral Pathol. 1981, 51, 320–328. [Google Scholar] [CrossRef]
- Fuks, A.B. Vital pulp therapy with new materials for primary teeth: New directions and treatment perspectives. Pediatr. Dent. 2008, 30, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Monsef, M.; Torabinejad, M. Sealing ability of a mineral trioxide aggregate for repair of lateral root perforations. J. Endod. 1993, 19, 541–544. [Google Scholar] [CrossRef]
- Witherspoon, D.E. Vital pulp therapy with new materials: New directions and treatment perspectives—Permanent teeth. Pediatr. Dent. 2008, 30, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Saunders, W.P. A prospective clinical study of periradicular surgery using mineral trioxide aggregate as a root-end filling. J. Endod. 2008, 34, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Húngaro Duarte, M.A.; de Oliveira El Kadre, G.D.; Vivan, R.R.; Guerreiro Tanomaru, J.M.; Filho, M.T.; de Moraes, I.G. Radiopacity of Portland Cement Associated With Different Radiopacifying Agents. J. Endod. 2009, 35, 737–740. [Google Scholar] [CrossRef]
- Laghios, C.D.; Benson, B.W.; Gutmann, J.L.; Cutler, C.W. Comparative radiopacity of tetracalcium phosphate and other root-end filling materials. Int. Endod. J. 2000, 33, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Chng, H.K.; Islam, I.; Yap, A.U.; Tong, Y.W.; Koh, E.T. Properties of a new root-end filling material. J. Endod. 2005, 31, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Danesh, G.; Dammaschke, T.; Gerth, H.U.; Zandbiglari, T.; Schafer, E. A comparative study of selected properties of ProRoot mineral trioxide aggregate and two Portland cements. Int. Endod. J. 2006, 39, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, J.; Gandolfi, M.G. Evaluation of the radiopacity of calcium silicate cements containing different radiopacifiers. Int. Endod. J. 2010, 43, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Min, K.S.; Chang, H.S.; Bae, J.M.; Park, S.H.; Hong, C.U.; Kim, E.C. The induction of heme oxygenase-1 modulates bismuth oxide-induced cytotoxicity in human dental pulp cells. J. Endod. 2007, 33, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Marciano, M.A.; Duarte, M.A.; Camilleri, J. Dental discoloration caused by bismuth oxide in MTA in the presence of sodium hypochlorite. Clin. Oral Investig. 2015, 19, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Coomaraswamy, K.S.; Lumley, P.J.; Hofmann, M.P. Effect of bismuth oxide radioopacifier content on the material properties of an endodontic Portland cement-based (MTA-like) system. J. Endod. 2007, 33, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Torabinejad, M.; Hong, C.U.; Pitt Ford, T.R.; Kettering, J.D. Cytotoxicity of four root end filling materials. J. Endod. 1995, 21, 489–492. [Google Scholar] [CrossRef]
- Ai, K.; Liu, Y.; Liu, J.; Yuan, Q.; He, Y.; Lu, L. Large-scale synthesis of Bi2S3 nanodots as a contrast agent for in vivo X-ray computed tomography imaging. Adv. Mater. 2011, 23, 4886–4891. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hsieh, S.C.; Teng, N.C.; Kao, C.K.; Lee, S.Y.; Lin, C.K.; Yang, J.C. Radiopacity and cytotoxicity of Portland cement containing zirconia doped bismuth oxide radiopacifiers. J. Endod. 2014, 40, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, E.A.; Guerreiro-Tanomaru, J.M.; Tanomaru-Filho, M.; Duarte, M.A. Radiographic effect of different radiopacifiers on a potential retrograde filling material. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2009, 108, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Coomaraswamy, K.S.; Lumley, P.J.; Shelton, R.M.; Hofmann, M.P. Evaluation of Different Radiopacifiers for an MTA-like Dental Cement. Key Eng. Mater. 2008, 361–363, 885–889. [Google Scholar] [CrossRef]
- Kossler, W.; Fuchs, J. Bioceramics: Properties, Preparation and Applications; Nova Science Publishers Inc.: New York, NY, USA, 2009; p. 299. ISBN 978-1-60741-056-0. [Google Scholar]
- Silva, V.V.; Lameiras, F.S.; Lobato, Z.I. Biological reactivity of zirconia-hydroxyapatite composites. J. Biomed. Mater. Res. 2002, 63, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Covacci, V.; Bruzzese, N.; Maccauro, G.; Andreassi, C.; Ricci, G.A.; Piconi, C.; Marmo, E.; Burger, W.; Cittadini, A. In vitro evaluation of the mutagenic and carcinogenic power of high purity zirconia ceramic. Biomaterials 1999, 20, 371–376. [Google Scholar] [CrossRef]
- Sorokina, S.L.; Sleight, A.W. New phases in the ZrO2–Bi2O3 and HfO2–Bi2O3 systems. Mater. Res. Bull. 1998, 33, 1077–1081. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, S.S.; Kim, W.J. Sol-gel synthesis and properties of multiferroic BiFeO3. Mater. Lett. 2005, 59, 4006–4009. [Google Scholar] [CrossRef]
- Yan, F.; Li, J.; Zhang, J.J.; Liu, F.Q.; Yang, W.S. Preparation of Fe3O4 polystyrene composite particles from monolayer oleic acid modified Fe3O4 nanoparticles via miniemulsion polymerization. J. Nanopart. Res. 2009, 11, 289–296. [Google Scholar] [CrossRef]
- Chen, C.; Cheng, J.; Yu, S.G.; Che, L.J.; Meng, Z.Y. Hydrothermal synthesis of perovskite bismuth ferrite crystallites. J. Cryst. Growth. 2006, 291, 135–139. [Google Scholar] [CrossRef]
- Suárez-Gómez, A.; Sato-Berrú, R.; Toscano, R.A.; Saniger-Blesa, J.M.; Calderón-Piñar, F. On the synthesis and crystallization process of nanocrystalline PZT powders obtained by a hybrid sol-gel alkoxides route. J. Alloy. Compd. 2008, 450, 380–386. [Google Scholar] [CrossRef]
- Shogren, R.L. Modification of maize starch by thermal processing in glacial acetic acid. Carbohydr. Polym. 2000, 43, 309–315. [Google Scholar] [CrossRef]
- Jin, W.; Abothu, I.R.; Wang, R.; Chung, T.S. Sol-Gel Synthesis and Characterization of SrFeCo0.5O3.25-δ Powder. Ind. Eng. Chem. Res. 2002, 41, 5432–5543. [Google Scholar] [CrossRef]
- Yu, X.; An, X. Enhanced magnetic and optical properties of pure and (Mn, Sr) doped BiFeO3 nanocrystals. Solid State Commun. 2009, 149, 711–714. [Google Scholar] [CrossRef]
- Levin, E.M.; McDaniel, C.L. Heats of Transformations in Bismuth Oxide by Differential Thermal Analysis. J. Res. Natl. Bur. Stand. A 1965, 69A, 237–243. [Google Scholar] [CrossRef]
- Harwig, H.A.; Gerards, A.G. The polymorphism of bismuth sesquioxide. Thermochim. Acta 1979, 28, 121–131. [Google Scholar] [CrossRef]
- Fruth, V.; Popa, M.; Berger, D.; Ionica, C.M.; Jitianu, M. Phases investigation in the antimony doped Bi2O3 system. J. Eur. Ceram. Soc. 2004, 24, 1295–1299. [Google Scholar] [CrossRef]
- Zhou, Q.F.; Chan, H.L.W.; Choy, C.L. Synthesis and properties of ferroelectric SrBi2Ta2O9 powder and films prepared by a sol-gel process. J. Non-Cryst. Solids 1999, 254, 106–111. [Google Scholar] [CrossRef]
- Hower, J.C.; Suárez-Ruiz, I.; Mastalerz, M.; Cook, A.C. The investigation of chemical structure of coal macerals via transmitted-light FT-IR microscopy by X. Sun. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 67, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Lin, J.; Fang, J. Silica Spheres Coated with YVO4:Eu3+ Layers via Sol-Gel Process: A Simple Method to Obtain Spherical Core−Shell Phosphors. Chem. Mater. 2005, 17, 1783–1791. [Google Scholar] [CrossRef]
- Li, W. Facile synthesis of monodisperse Bi2O3 nanoparticles. Mater. Chem. Phys. 2006, 99, 174–810. [Google Scholar] [CrossRef]
- Sood, K.; Singh, K.; Pandey, O.P. Synthesis and characterization of Bi-doped zirconia for solid electrolyte. Ionics 2010, 16, 549–554. [Google Scholar] [CrossRef]
- Levin, E.M.; McDaniel, C.L. Polymorphism of Bismuth Sesquioxide. II. Effect of Oxide Additions on the Polymorphism of Bi2O3. J. Res. Natl. Bur. Stand. A Phys. Chem. 1964, 68A, 197–206. [Google Scholar] [CrossRef]
- Tyagi, B.; Sidhpuria, K.; Shaik, B.; Jasra, R.V. Synthesis of Nanocrystalline Zirconia Using Sol-Gel and Precipitation Techniques. Ind. Eng. Chem. Res. 2006, 45, 8643–8650. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | Means and Standard Deviations of Radiopacity * (mmAl) | |||||||
|---|---|---|---|---|---|---|---|---|
| PC | PC/Bi2O3 | 0 | 0.2 | 0.4 | 0.6 | 0.8 | 1.0 | |
| 0.96 ± 0.07 | 4.69 ± 0.23 | |||||||
| 500 | 4.68 ± 0.13 | 4.90 ± 0.23 | 4.48 ± 0.23 | 3.89 ± 0.21 | 3.95 ± 0.21 | 3.75 ± 0.17 | ||
| 600 | 4.17 ± 0.10 | |||||||
| 650 | 4.61 ± 0.17 | |||||||
| 700 | 4.73 ± 0.15 | |||||||
| 750 | 4.99 ± 0.16 | 5.83 ± 0.22 | 4.45 ± 0.20 | 4.39 ± 0.22 | 4.01 ± 0.20 | 3.72 ± 0.16 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.-S.; Chen, S.-H.; Lai, F.-C.; Chen, C.-Y.; Hsieh, M.-Y.; Chang, W.-J.; Yang, J.-C.; Lin, C.-K. Sintering Temperature-Dependence on Radiopacity of Bi(2−x) ZrxO(3+x/2) Powders Prepared by Sol-Gel Process. Materials 2018, 11, 1685. https://doi.org/10.3390/ma11091685
Chen M-S, Chen S-H, Lai F-C, Chen C-Y, Hsieh M-Y, Chang W-J, Yang J-C, Lin C-K. Sintering Temperature-Dependence on Radiopacity of Bi(2−x) ZrxO(3+x/2) Powders Prepared by Sol-Gel Process. Materials. 2018; 11(9):1685. https://doi.org/10.3390/ma11091685
Chicago/Turabian StyleChen, May-Show, Shih-Hsun Chen, Fu-Chih Lai, Chin-Yi Chen, Ming-Yuan Hsieh, Wei-Jen Chang, Jen-Chang Yang, and Chung-Kwei Lin. 2018. "Sintering Temperature-Dependence on Radiopacity of Bi(2−x) ZrxO(3+x/2) Powders Prepared by Sol-Gel Process" Materials 11, no. 9: 1685. https://doi.org/10.3390/ma11091685