Half-Metallic Property Induced by Double Exchange Interaction in the Double Perovskite Bi2BB′O6 (B, B′ = 3d Transitional Metal) via First-Principles Calculations

Abstract
:1. Introduction
2. Materials and Methods
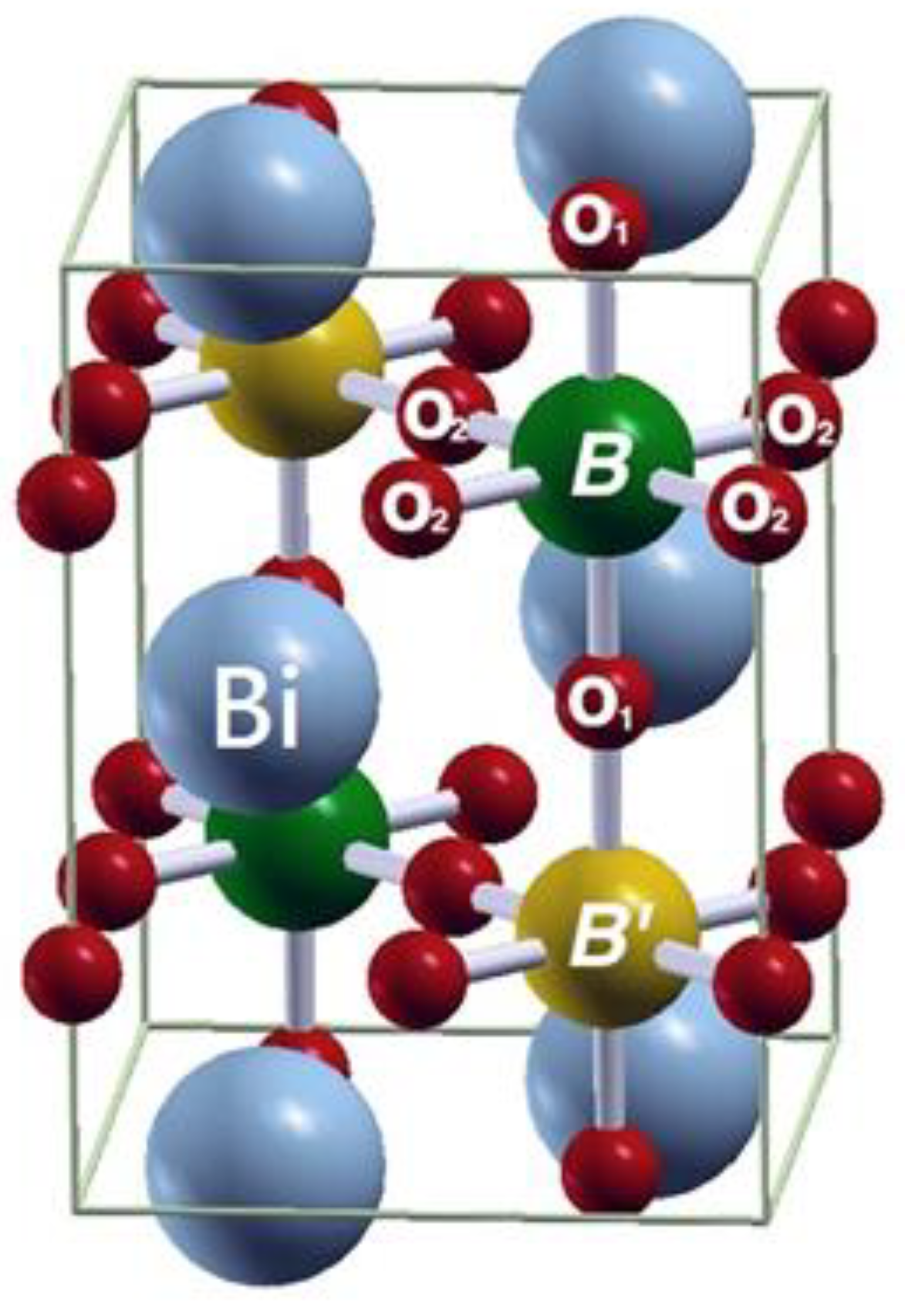
2.1. Structural Optimization
2.2. Calculation Method and Procedure
3. Results and Discussion
3.1. FM-HM Compounds: Bi2CrCoO6, Bi2CrNiO6, and Bi2FeNiO6
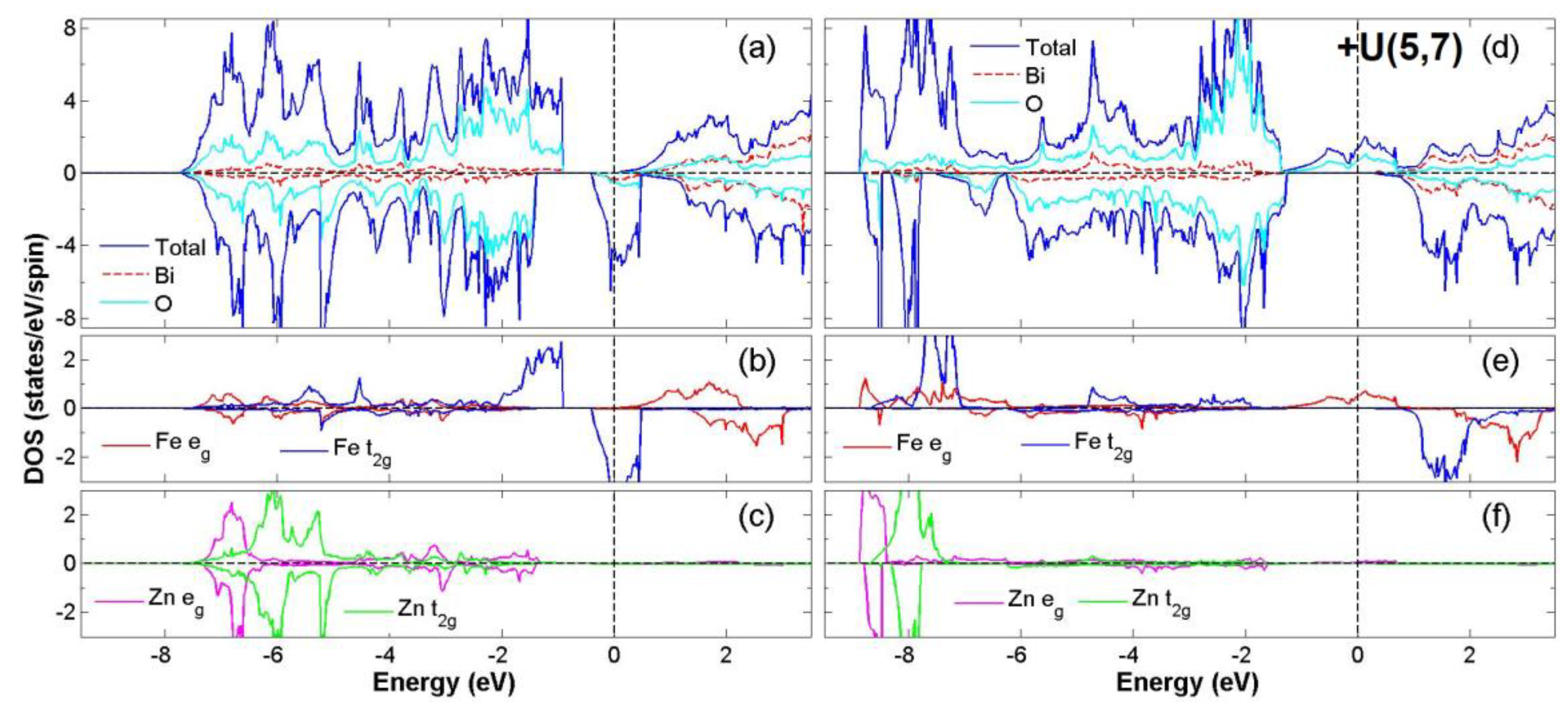
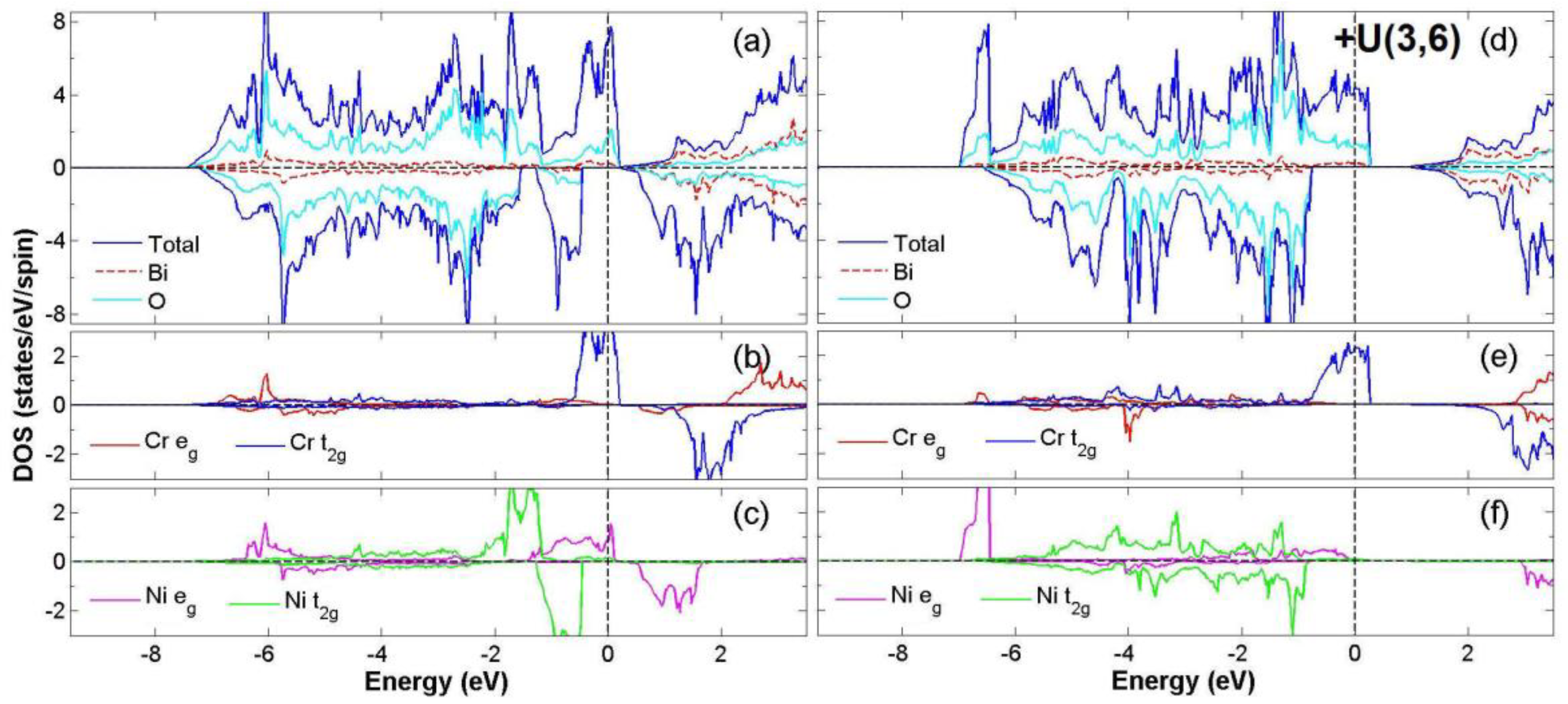
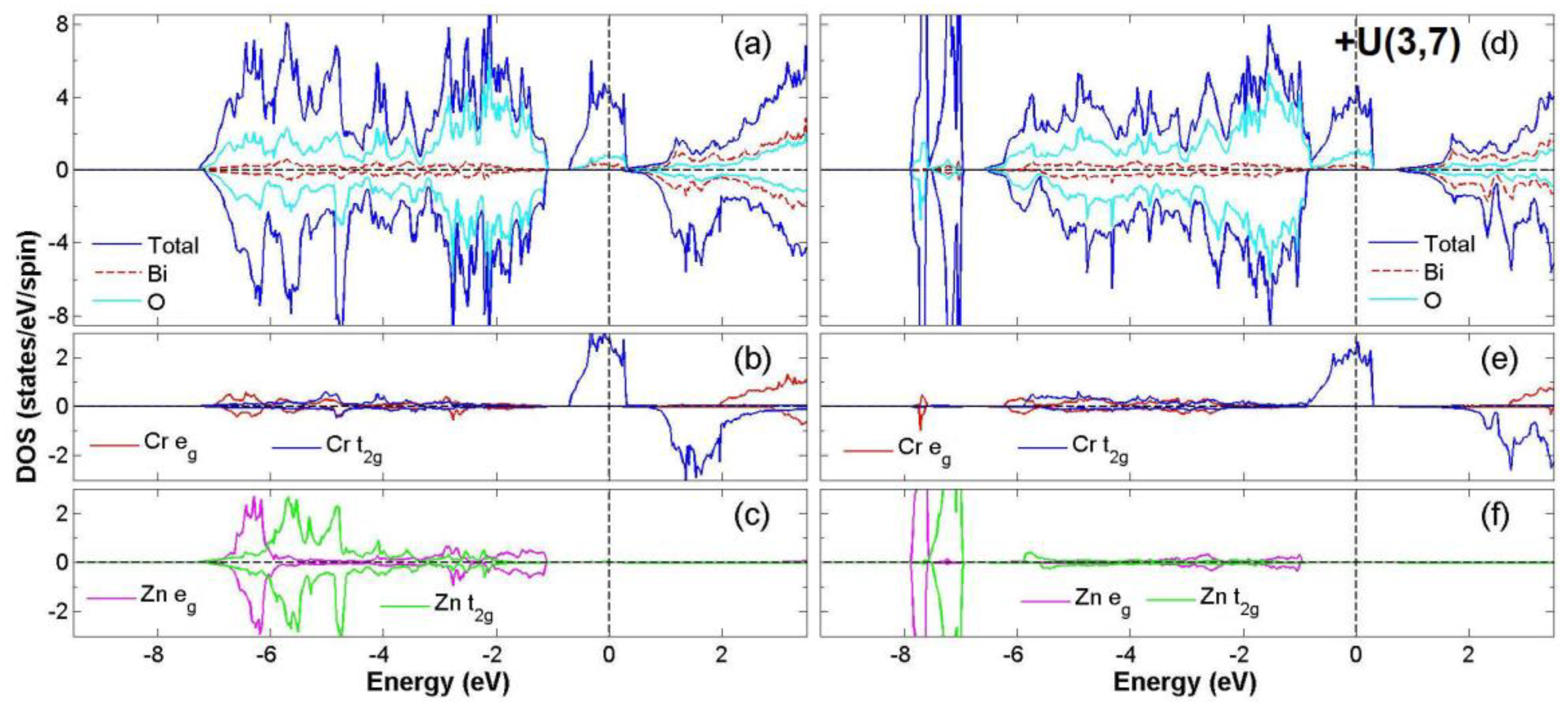
3.2. FiM-HM Compounds: Bi2FeZnO6, Bi2CrZnO6 and Bi2CoZnO6
3.3. AF Compounds: Bi2CrCoO6 and Bi2FeNiO6
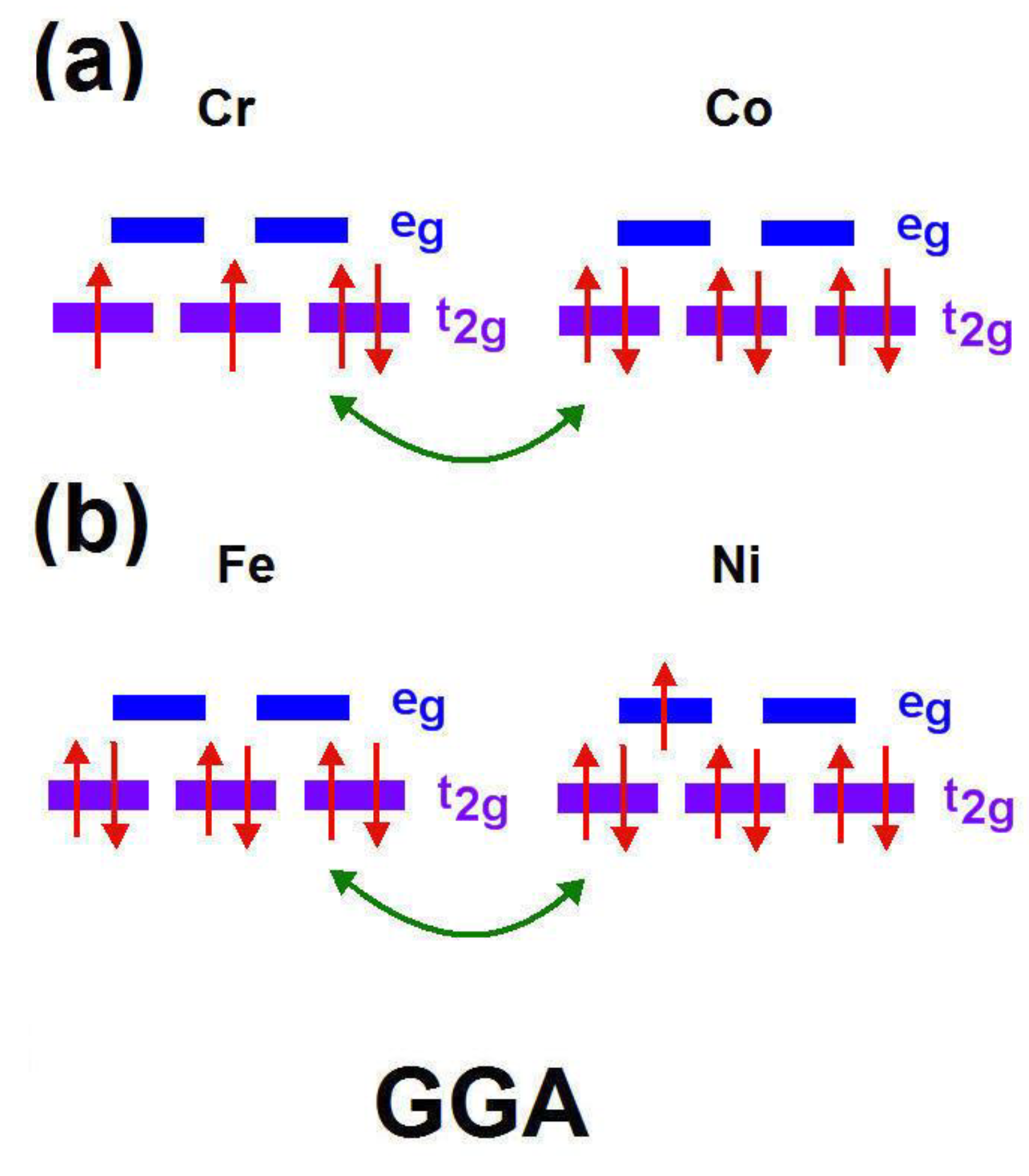
3.4. Double Exchange Interaction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, J.H.; Vescovo, E.; Kim, H.J.; Kwon, C.; Ramesh, R.; Venkatesan, T. Direct evidence for a half-metallic ferromagnet. Nature 1998, 392, 794–796. [Google Scholar] [CrossRef]
- Pickett, W.E.; Moodera, J.S. Half metallic magnets. Phys. Today 2001, 54, 39–44. [Google Scholar] [CrossRef]
- Kobayashi, K.I.; Kimura, T.; Sawada, H.; Terakura, K.; Tokura, Y. Room-temperature magnetoresistance in an oxide material with an ordered double-perovskite structure. Nature 1998, 395, 677–680. [Google Scholar] [CrossRef]
- Schwarz, K.J. CrO2 predicted as a half-metallic ferromagnet. Phys. F Met. Phys. 1986, 16, 211–216. [Google Scholar] [CrossRef]
- Jeng, H.T.; Guo, G.Y. First-principles investigations of orbital magnetic moments and electronic structures of the double perovskites Sr2FeMoO6, Sr2FeReO6, and Sr2CrWO6. Phys. Rev. B 2003, 67, 094438. [Google Scholar] [CrossRef]
- Chan, T.S.; Liu, R.S.; Guo, G.Y.; Hu, S.F.; Lin, J.G.; Chen, J.M.; Chang, C.R. Effects of B′-site transition metal on the properties of double perovskites Sr2FeMO6 (M = Mo, W): B′ 4d–5d system. Solid State Commun. 2005, 133, 265–270. [Google Scholar] [CrossRef]
- Wu, H. Electronic structure study of double perovskites A2FeReO6 (A = Ba, Sr, Ca) and Sr2MMoO6 (M = Cr, Mn, Fe, Co) by LSDA and LSDA + U. Phys. Rev. B 2001, 64, 125126. [Google Scholar] [CrossRef]
- Meetei, O.N.; Erten, O.; Mukherjee, A.; Randeria, M.; Trivedi, N.; Woodward, P. Theory of half-metallic double perovskites. I. Double exchange mechanism. Phys. Rev. B 2013, 87, 165104. [Google Scholar] [CrossRef]
- Erten, O.; Meetei, O.N.; Mukherjee, A.; Randeria, M.; Trivedi, N.; Woodward, P. Theory of half-metallic double perovskites. II. Effective spin Hamiltonian and disorder effects. Phys. Rev. B 2013, 87, 165105. [Google Scholar] [CrossRef]
- Wang, Y.K.; Lee, P.H.; Guo, G.Y. Half-metallic antiferromagnetic nature of La2VTcO6 and La2VCuO6 from ab initio calculations. Phys. Rev. B 2009, 80, 224418. [Google Scholar] [CrossRef]
- Coey, J.M.D.; Viret, M.; von Molnar, S. Mixed-valence manganites. Adv. Phys. 1999, 48, 167–293. [Google Scholar] [CrossRef]
- Kronik, L.; Jain, M.; Chelikowsky, J.R. Electronic structure and spin polarization of MnxGa1−xN. Phys. Rev. B 2002, 66, 041203. [Google Scholar] [CrossRef]
- Park, M.S.; Kwon, S.K.; Toun, S.J.; Min, B.I. Half-metallic electronic structures of giant magnetoresistive spinels: Fe1−xCuxCr2S4 (x = 0.0, 0.5, 1.0). Phys. Rev. B 1999, 59, 10018. [Google Scholar] [CrossRef]
- Shirai, M.; Ogawa, T.; Kitagawa, I.; Suzuki, N. Band structures of zinc-blende-type MnAs and (MnAs)1(GaAs)1 superlattice. J. Magn. Magn. Mater. 1998, 177, 1383–1384. [Google Scholar] [CrossRef]
- Park, J.H.; Kwon, S.K.; Min, B.I. Electronic structures of III–V based ferromagnetic semiconductors: Half-metallic phase. Phys. B 2000, 281, 703–704. [Google Scholar] [CrossRef]
- Chen, G.; Balents, L. Spin-orbit coupling in d2 ordered double perovskites. Phys. Rev. B 2011, 84, 094420. [Google Scholar] [CrossRef]
- Fuh, H.R.; Weng, K.C.; Liu, Y.P.; Wang, Y.K. Theoretical prediction of a new type of half-metallic materials in double perovskite Pb2BB′O6 (B, B′ = 3d transition metal) via First-Principle calculations. In Spin; World Scientific Publishing Company: Singapore, 2014; Volume 4, p. 1450001. [Google Scholar]
- Li, Z.; Cho, Y.; Li, X.; Li, X.; Aimi, A.; Inaguma, Y.; Alonso, J.A.; Fernandez-Diaz, M.T.; Yan, J.; Downer, M.C.; et al. New mechanism for ferroelectricity in the perovskite Ca2−xMnxTi2O6 synthesized by spark plasma sintering. J. Am. Chem. Soc. 2018, 140, 2214–2220. [Google Scholar] [CrossRef]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef]
- Seo, J.; Noh, J.H.; Seok, S.I. Rational strategies for efficient perovskite solar cells. Acc. Chem. Res. 2016, 49, 562–572. [Google Scholar] [CrossRef]
- Zuo, C.; Bolink, H.J.; Han, H.; Huang, J.; Cahen, D.; Ding, L. Advances in perovskite solar cells. Adv. Sci. 2016, 3, 1500324. [Google Scholar] [CrossRef]
- Yang, W.S.; Park, B.-W.; Jung, E.H.; Jeon, N.J.; Kim, Y.C.; Lee, D.U.; Shin, S.S.; Seo, J.; Kim, E.K.; Noh, J.H.; et al. Iodide management in formamidinium-lead-halide–based perovskite layers for efficient solar cells. Science 2017, 356, 1376–1379. [Google Scholar] [CrossRef] [PubMed]
- Unger, E.L. The PV-Researcher’s Siren: Hybrid metal halide perovskites. Curr. Opin. Green Sustain. Chem. 2017, 4, 72–76. [Google Scholar] [CrossRef]
- Zhou, X.; Jankowska, J.; Dong, H.; Prezhdo Oleg, V. Recent theoretical progress in the development of perovskite photovoltaic materials. J. Energy Chem. 2018, 27, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yin, W. Recent progress in Pb-free stable inorganic double halide perovskites. J. Semicond. 2018, 39, 071003. [Google Scholar] [CrossRef]
- Kato, H.; Okuda, T.; Okimoto, Y.; Tomioka, Y. Structural and electronic properties of the ordered double perovskites A2MReO6 (A = Sr, Ca; M = Mg, Sc, Cr, Mn, Fe, Co, Ni, Zn). Phys. Rev. B 2004, 69, 184412. [Google Scholar] [CrossRef]
- Tang, C.Q.; Zhang, Y.; Dai, J. Electronic and magnetic structure studies of double perovskite Sr2CrReO6 by first-principles calculations. Solid State Commun. 2005, 133, 219–222. [Google Scholar] [CrossRef]
- Philipp, J.B.; Majewski, P.; Alff, L.; Erb, A.; Gross, R.; Graf, T.; Brandt, M.S.; Simon, J.; Walther, T.; Mader, W.; et al. Structural and doping effects in the half-metallic double perovskite A2CrWO6 (A = Sr, Ba, and Ca). Phys. Rev. B 2003, 68, 144431. [Google Scholar] [CrossRef]
- Liu, Y.P.; Fuh, H.R.; Xiao, Z.R.; Wang, Y.K. Theoretical prediction of half-metallic materials in double perovskites Sr2Cr(Co)B′O6 (B′ = Y, La, Zr, and Hf) and Sr2V(Fe)B′O6 (B′ = Zr and Hf). J. Alloys Compd. 2014, 586, 289–294. [Google Scholar] [CrossRef]
- Nakamura, T.; Choy, J.H. Determination of ionic valency pairs via lattice constants in ordered perovskites (ALa)(Mn2+Mo5+)O6 (A = Ba, Sr, Ca) with applications to (ALa)(Fe3+Mo4+)O6, Ba2(Bi3+Bi5+)O6 and Ba2(Bi3+Sb5+)O6. J. Solid State Chem. 1977, 20, 233–244. [Google Scholar] [CrossRef]
- Zener, C. Interaction between the d-Shells in the Transition Metals. II. Ferromagnetic Compounds of Manganese with Perovskite Structure. Phys. Rev. 1951, 82, 403. [Google Scholar] [CrossRef]
- Anderson, P.W.; Hasegawa, H. Considerations on double exchange. Phys. Rev. 1955, 100, 675. [Google Scholar] [CrossRef]
- Pradhan, K.; Jena, P. Double exchange mediated ferromagnetic coupling between Co atoms in dicobalt complex. Appl. Phys. Lett. 2011, 99, 153105. [Google Scholar] [CrossRef] [Green Version]
- Nolting, W.; Ramakanth, A. Quantum Theory of Magnetism; Springer: Heidealberg, Germany; Dordrecht, The Netherlands; London, UK; New York, NY, USA, 2009; ISBN 978-3-540-85415-9. [Google Scholar] [CrossRef]
- Solovyev, I.V.; Terakura, K. Zone Boundary Softening of the Spin-Wave Dispersion in Doped Ferromagnetic Manganites. Phys. Rev. Lett. 1999, 82, 2959. [Google Scholar] [CrossRef]
- Solovyev, I.V.; Terakura, K. Electronic Structure and Magnetism of Complex Materials; Singh, D.J., Papaconstantopoulos, D.A., Eds.; Springer: Berlin, Germany, 2003. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Solovyev, I.V.; Dederichs, P.H.; Anisimov, V.I. Corrected atomic limit in the local-density approximation and the electronic structure of d impurities in Rb. Phys. Rev. B 1994, 50, 16861–16871. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bi2BB′O6 | a | c/a | V0 (Å3/f.u.) | O1z | O2x | O2y |
|---|---|---|---|---|---|---|
| CrCo | 5.419 | 1.413 | 112.461 | 0.2528 | 0.2471 | 0.2471 |
| CrNi | 5.453 | 1.415 | 114.724 | 0.2473 | 0.2526 | 0.2526 |
| FeNi | 5.421 | 1.413 | 112.606 | 0.2453 | 0.2546 | 0.2546 |
| FeZn | 5.464 | 1.414 | 115.332 | 0.2399 | 0.2600 | 0.2600 |
| CrZn | 5.505 | 1.416 | 118.095 | 0.2414 | 0.2587 | 0.2587 |
| CoZn | 5.450 | 1.415 | 114.467 | 0.2400 | 0.2600 | 0.2600 |
| Materials Bi2BB′O6 | (UB,UB′) | Spin Magnetic Moment (μB/f.u.) | d Orbital Electrons↑/↓ | Band Gap (eV) | ∆E (meV/f.u.) FM(FiM) | |||
|---|---|---|---|---|---|---|---|---|
| mB | mB′ | mtot | B | B′ | ||||
| CrCo | (0, 0) | 2.573 | 0.162 | 3.000 | 3.465/0.927 | 3.749/3.575 | 0.00/0.40 | −25 |
| (3, 6) | 2.852 | 3.071 | 6.792 | 3.574/0.804 | 5.108/2.044 | 0.00/0.00 | 903 | |
| CrNi | (0, 0) | 2.304 | 1.267 | 4.000 | 3.304/1.076 | 4.767/3.496 | 0.00/0.65 | −97 |
| (3, 6) | 2.303 | 1.703 | 4.000 | 3.272/1.036 | 5.019/3.320 | 0.00/1.73 | −112 | |
| FeNi | (0, 0) | 2.146 | 1.224 | 4.000 | 4.143/2.054 | 4.749/3.528 | 0.15/0.00 | −43 |
| (5, 6) | 3.887 | 1.644 | 6.000 | 4.919/1.094 | 4.939/3.341 | 0.00/1.68 | 34 | |
| FeZn | (0, 0) | 1.694 | −0.022 | 2.000 | 3.950/2.269 | 4.968/4.984 | 0.97/0.00 | −58 |
| (5, 7) | 3.66 | 0.015 | 4.000 | 4.848/1.223 | 5.022/5.051 | 0.00/1.52 | 1125 | |
| CrZn | (0, 0) | 1.866 | −0.013 | 2.000 | 3.111/1.274 | 4.981/4.988 | 0.00/1.30 | −60 |
| (3, 7) | 2.073 | −0.008 | 2.000 | 3.202/1.165 | 5.050/5.054 | 0.00/1.57 | −73 | |
| CoZn | (0, 0) | 0.758 | −0.013 | 1.000 | 3.995/3.242 | 4.966/4.976 | 0.85/0.00 | −51 |
| (6, 7) | 3.463 | 0.105 | 5.000 | 5.229/1.782 | 5.080/5.025 | 0.65/0.75 | 99 | |
| CrCo(AF) | (0, 0) | 2.579 | 0.057 | 0.000 | 3.467/0.925 | 3.692/3.631 | 0.00/0.00 | – |
| (3, 6) | 2.799 | 3.194 | 0.000 | 3.561/0.816 | 5.129/1.952 | 0.53/0.53 | – | |
| FeNi(AF) | (0, 0) | 2.805 | 0.836 | 0.000 | 4.439/1.675 | 4.567/3.735 | 0.00/0.00 | – |
| (5, 6) | 3.847 | 1.517 | 0.000 | 4.894/1.089 | 4.912/3.410 | 0.00/0.0 | – | |
| Materials Bi2BB′O6 | (UB,UB′) | Final State | E(eV/f.u.) | Materials Bi2BB′O6 | (UB,UB′) | Final State | E(eV/f.u.) (eV) |
|---|---|---|---|---|---|---|---|
| CrCo | (0, 0) | AF | −65.880 | FeZn | (0, 0) | AF | −57.804 |
| (3, 6) | AF | −61.827 | – | (5, 7) | AF | −56.040 | |
| (0, 0) | FM | −65.905 | – | (0, 0) | FiM | −57.862 | |
| (3, 6) | FM | −60.924 | – | (5, 7) | FM | −54.915 | |
| CrNi | (0, 0) | AF | −63.883 | CrZn | (0, 0) | AF | −60.725 |
| (3, 6) | AF | −60.877 | – | (3, 7) | AF | −58.949 | |
| (0, 0) | FM | −63.980 | – | (0, 0) | FiM | −60.785 | |
| (3, 6) | FM | −60.989 | – | (3, 7) | FiM | −59.022 | |
| FeNi | (0, 0) | AF | −61.036 | CoZn | (0, 0) | AF | −55.693 |
| (5, 6) | AF | −57.053 | – | (6, 7) | AF | −51.111 | |
| (0, 0) | FM | −61.079 | – | (0, 0) | FiM | −55.744 | |
| (5, 6) | FM | −57.019 | – | (6, 7) | FM | −51.012 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-Z.; Hu, C.-Y.; Lee, P.-H.; Yan, A.Z.-Z.; Wu, W.-F.; Chen, Y.-F.; Wang, Y.-K. Half-Metallic Property Induced by Double Exchange Interaction in the Double Perovskite Bi2BB′O6 (B, B′ = 3d Transitional Metal) via First-Principles Calculations. Materials 2019, 12, 1844. https://doi.org/10.3390/ma12111844
Lin H-Z, Hu C-Y, Lee P-H, Yan AZ-Z, Wu W-F, Chen Y-F, Wang Y-K. Half-Metallic Property Induced by Double Exchange Interaction in the Double Perovskite Bi2BB′O6 (B, B′ = 3d Transitional Metal) via First-Principles Calculations. Materials. 2019; 12(11):1844. https://doi.org/10.3390/ma12111844
Chicago/Turabian StyleLin, Hong-Zong, Chia-Yang Hu, Po-Han Lee, Albert Zhong-Ze Yan, Wen-Fang Wu, Yang-Fang Chen, and Yin-Kuo Wang. 2019. "Half-Metallic Property Induced by Double Exchange Interaction in the Double Perovskite Bi2BB′O6 (B, B′ = 3d Transitional Metal) via First-Principles Calculations" Materials 12, no. 11: 1844. https://doi.org/10.3390/ma12111844