3.1. WAXS Results
The changes in the WAXS profiles were measured in the isothermal crystallization process at 110 °C from the melt (200 °C).
Figure 1 shows the WAXS profiles for the D1.4 neat and D1.4/SFN(1.0) specimens as a function of time. Here,
q denotes the magnitude of the scattering vector, as defined by
q = (4π/
λ) sin(
θ/2), with
λ and
θ being the wavelength of X-ray and the scattering angle, respectively. The indexing of the observed reflections is based on the crystal structure of PLLA reported in the references [
3,
4,
5].
As shown in
Figure 1, in the early stage, there is no crystalline peak, which shows the presence of 100% amorphous phase. As time goes on, a crystalline peak appears (which has been shown by the red arrow in
Figure 1). The induction period (
t0) of the crystallization is evaluated from the first detection of the crystalline peak. It can be seen from
Figure 1 that the loading 1% SFN caused the reduction of the induction period from 90 s to 40 s, which shows the enhancement of the crystallizability by SFN. The time evolution of the degree of crystallinity was calculated from the WAXS profiles by using the following equation:
Here,
ΣAc is the summation of the peak area of the crystalline peaks, and
Aa is the peak area of the amorphous halo. The peak decomposition was conducted, and the degree of crystallinity (
ϕWAXS) was evaluated. The calculated
ϕWAXS is plotted as function of time in
Figure 2. As can be seen from
Figure 2, the final degree of the crystallinity has been increased by the inclusion of 1% SFN. Judging from the slope of the curve at
t =
t0.5 (where
t0.5 is the crystallization half-time at the 50% of the final crystallinity attained) in
Figure 2, it can be considered that the crystallizabilty of PLLA is increased by adding 1% SFN.
Table 2 summarizes the induction period, the final degree of crystallinity and the crystallization half-time of all the specimens, calculated from the results of
Figure 2. It is clearly observed that
t0.5 is also decreased by loading of SFN, indicating the acceleration of the crystallization rate.
Furthermore, it is important to check the effect of SFN on the formation of the crystal polymorph (α and α’ phases). For this purpose, the evaluation of the fraction of α and α’ phases formed during the isothermal crystallization at 110 °C was conducted by analyzing the (200)/(110) reflection. Since there observed a small shoulder around
q = 12.2 nm
−1, we tried to decompose the shoulder peak (α phase) from the main peak (α’ phase).
Figure 3 shows an example of the peak decomposition in the range of 11 <
q < 13 nm
−1, ensuring the perfect peak decomposition.
Figure 4 shows the changes in the peak positions of the α’ and α phases. The peak positions of both of the phases are continuously increasing as the crystallization process proceeds. These results show that loosely packed crystals are initially formed and gradually densified as a function of time. There was almost no effect of SFN on the overall behaviors of the changes in the peak position.
On the basis of the area of the decomposed (200)/(110) peaks, the fraction of α and α’ crystals are evaluated as
where
Ax denotes the peak area of the decomposed
x reflection.
Figure 5a,b shows the peak area of the α and α’ phases as a function of time. Firstly, the α’ peak appeared, showing that loosely packed crystals are initially developed. After
t = 5 min for the D1.4 neat or
t = 3.5 min for the D1.4/SFN(1.0) specimen, the α phase starts appearing. The peak areas of both phases are increasing as a function of time.
Figure 5c,d shows the fraction of the α and α’ phases evaluated by using Equations (2) and (3). As seen in
Figure 5c, the fraction of the α phase is increasing in the early stage because of the successive formation of the α phase in the preceding α’ phase crystallites. However, the fraction of the α phase was leveled off at 3.8% to 4%. This means that the increasing behaviors for both phases are similar to each other, as can be seen in
Figure 5a,b. Finally, it can be stated that the SFN may decrease the α fraction slightly. A similar result has been found for the case of loading a special plasticizer (organic acid monoglyceride; OMG) [
13], where the reduction of the α phase is favorable because of the unfavorable nature of the lower impact strength of the α phase (more brittle than the α’ phase). Furthermore, we discuss whether the α phase is formed directly from the melt or due to the transition of the α’ phase. It has been reported [
5] that the transition of α’-to-α phase takes place in heating prior to the melting. Since the temperature is kept constant (isothermal crystallization), it is needless to consider the α’-to-α crystal transition. Moreover, as shown in
Figure 5b, the amount (peak area) of the α’ phase is not decreased even after the appearance of the α phase, which suggests that the α crystals are directly formed from melt instead of α’-to-α phase transition.
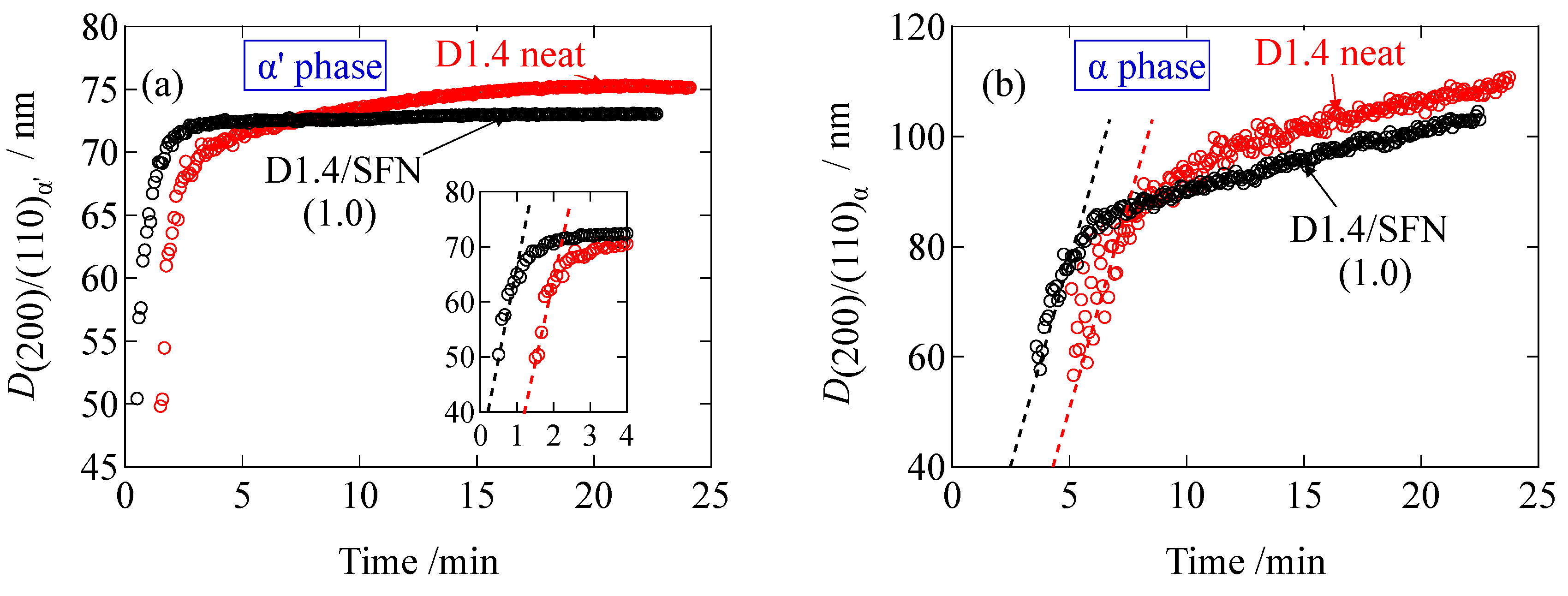
The average crystallite size (
Dhkl) in the direction normal to the (
hkl) plane was evaluated by the Scherrer’s equation,
where
K is a constant (0.9) and
λ is the wavelength of the incident X-ray.
β is a full-width at half maximum (FWHM) in the unit of radian, and
θ is the scattering angle.
As seen in
Figure 6, the crystallite size is increasing as a function of time during the crystallization. The final value of the crystallite size for the D1.4/SFN(1.0) specimen is smaller than the D1.4 neat specimen. This result indicates that the presence of SFN decreases the final size of the crystallites of PLLA. The slope of the plots in
Figure 6 can be considered as the crystallite growth rate. As can be seen in
Figure 6a,b, the initial slope is almost constant, indicating that the SFN does not accelerate the growth of the crystallites. However, the SFN shortens the induction period (see also
Figure 6a,b) as the evolution time is much shortened by SFN, suggesting that the SFN can enhance only the nucleation rate and not the crystallite growth rate. Similar results have been reported in our previous study for the spherulite growth based on the POM observation [
11], where it was reported that the spherulite growth was unaffected by the inclusion of SFN, while the induction period was reduced. Finally, it should be noted that
D(200)/(110) stands for the lateral size of the lamellar crystallites. The initial slope of the plot in
Figure 6a gives 30.0 nm/min of the growth rate of the lateral direction of the crystalline lamella (α’ phase), while the slope in
Figure 6b gives the growth rate of 14.8 nm/min for the α phase. These values are much larger as compared to the growth rate of the lamellar thickness, which is 0.3 nm/min (as shown later). The much slower growth of the lamellar thickness is quite reasonable.
The kinetics of isothermal crystallization are described by the well-known Avrami theory [
14,
15,
16], according to which the degree of crystallinity as a function of time [
ϕ(
t)] can be expressed as:
Here,
ϕ∞ denotes the degree of crystallinity after the complete crystallization,
t0 is the induction period for the crystallization,
n is the Avrami exponent which represents the dimensionality of the growing crystallites and
k is the crystallization rate constant, which contains the contributions from the nucleation and the growth of crystal. The Avrami plots for the D1.4 neat, D1.4/SFN(1.0), D0.5 neat and D0.5/SFN(1.0) specimens are shown in
Figure 7. Here, the initial slope of the Avrami plots was different from that in the later stage, suggesting the change in the mode of the crystal growth. The crossover time is earlier for the D0.5 neat specimen than the D1.4 neat specimen. However, in the case of the D0.5/SFN(1.0) specimen, such crossover behavior was not observed. The similar tendency of the Avrami plots has been found in our previous study [
11] in which the Avrami plots are made on the basis of the results of the DSC measurements. In the early stage of the crystallization, the Avrami exponent (
n) for the D1.4 neat specimen and for the D0.5 neat specimen was
n = 1.7 for both, suggesting the one-dimensional crystal growth because of the homogenous nucleation for the D1.4 neat and the D1.4/SFN(1.0) specimens in the early stage of the crystallization (
t < 3.9 min or
t < 14 min), respectively, for those specimens as reported in our previous paper [
11]. However, in the later stage of the crystallization, the Avrami exponent (
n) was increased from 2.2 (D1.4 neat) to 2.8 (D1.4/SFN(1.0)) by the loading of SFN, which suggests the change of the dimensionality of the crystal growth from 2 dimensional to 3-dimensional by SFN because of the heterogenous nucleation. Such a crossover is also confirmed in the D0.5 neat specimen. If we compare the later stage of the crystallization for the D0.5 neat and D0.5/SFN(1.0) specimens, the Avrami exponent (
n) is also increased from
n = 2.2 to 3.0. Such a change of the dimensionality of the crystal growth was observed for the Avrami plots based on the DSC results in our previous study [
11] with almost similar values of the Avrami exponent (
n). These results suggest that the SFN increases the dimensionality of the crystal growth, and hence the crystallization rate (crystallinity increasing rate) is accelerated.
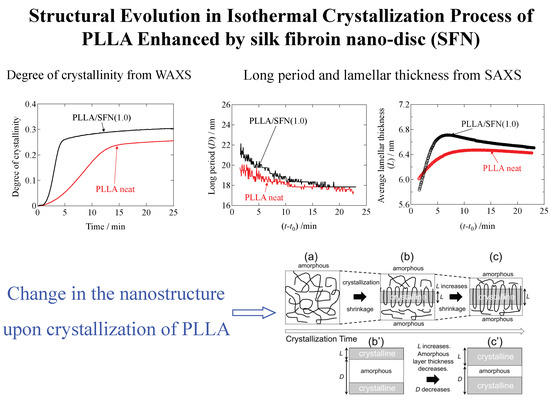
3.2. SAXS Results
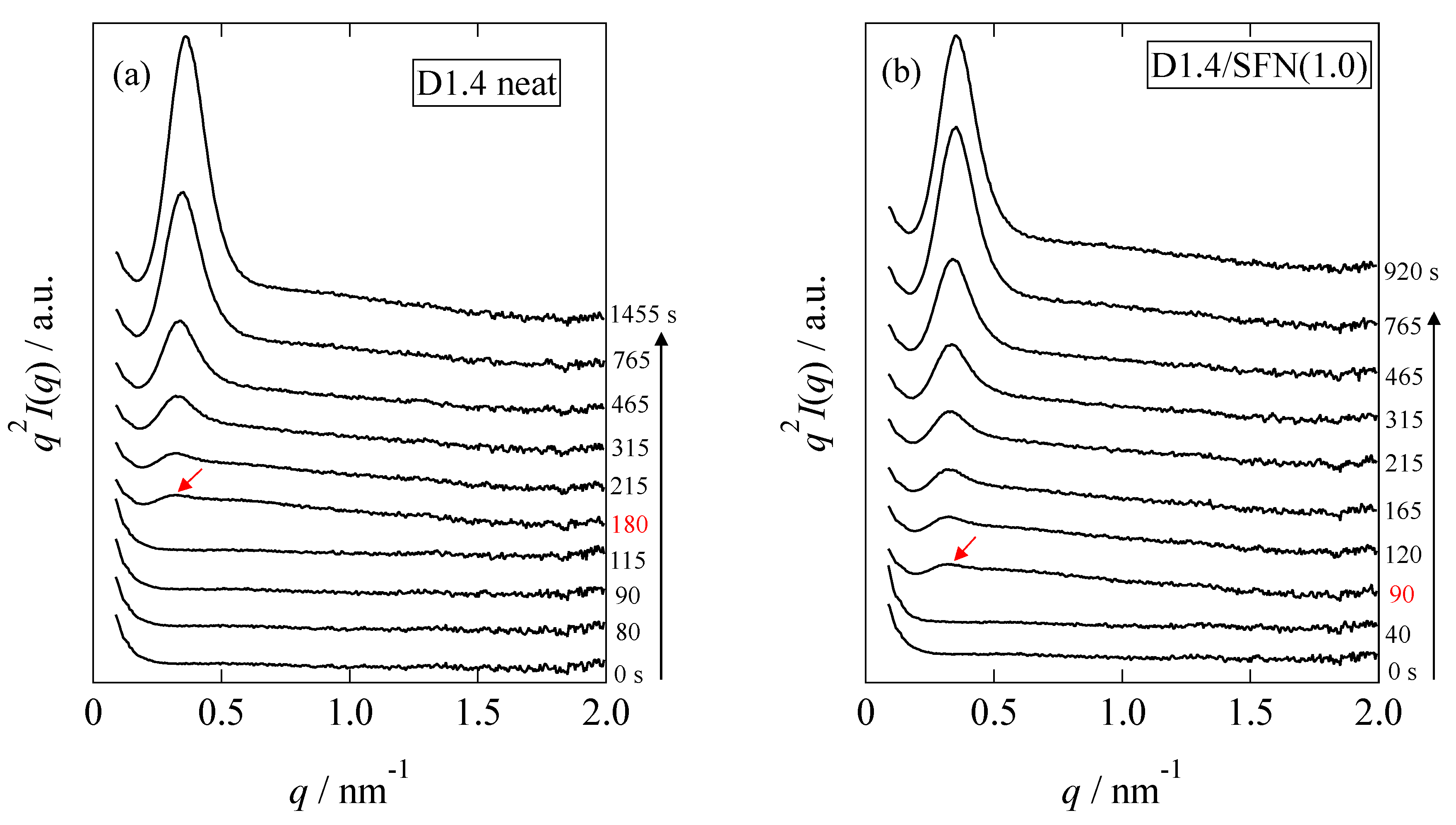
Figure 8 shows the changes in the Lorentz-corrected SAXS profiles as a function of time for the D1.4 neat and D1.4/SFN(1.0) specimens. Here, the scattering intensity,
I(
q), is corrected as
q2I(
q) by multiplying
q2. In the early stage of the crystallization, there was no observation of the peak. At
t = 180 s for the D1.4 neat or
t = 90 s for the D1.4/SFN(1.0) specimen, a clear scattering peak was observed, which indicates the development of the lamellar stacking with sandwiching the amorphous layers. It is notable to observe that the WAXS peak appears earlier than the SAXS peak (
Figure 1 and
Figure 8). Such a result indicates that during the early stage of crystallization, the lamellar stacks are incomplete by noting
t0 = 90 s and 40 s for the D1.4 neat and D1.4/SFN(1.0) specimens (see
Figure 1). In other words, single lamellae (without stacking) are generated in the early stage of the crystallization.
The intensity of the peak observed at
q = 0.29 nm
−1 increases as a function of time. From the peak position (
q*), the long period (
D) of the lamellar stacks was evaluated as
D = 2π/
q*. As seen in
Figure 8, the SAXS peak moves towards the higher
q as the crystallization proceeds. Increase in
q suggests the decrease in
D.
As shown in
Figure 9a, the repeating distance of the lamellar stacks (
D) decreases as a function of the crystallization time. This result seemed to be opposed to the process of crystallization. In order to understand this behavior, we evaluated the average thickness of the crystalline lamella (
L). To evaluate
L, the correlation function
γ(
r) was calculated from the 1d- SAXS profile through the following equation (inverse Fourier transform method):
Here,
γ(
r) is the correlation function and
r is the distance in the real space.
Figure 9 shows thus-evaluated
L and
D and the ratio (
L/
D) as a function of time during the isothermal crystallization upon T-jump from 200 °C to 110 °C. As a result (
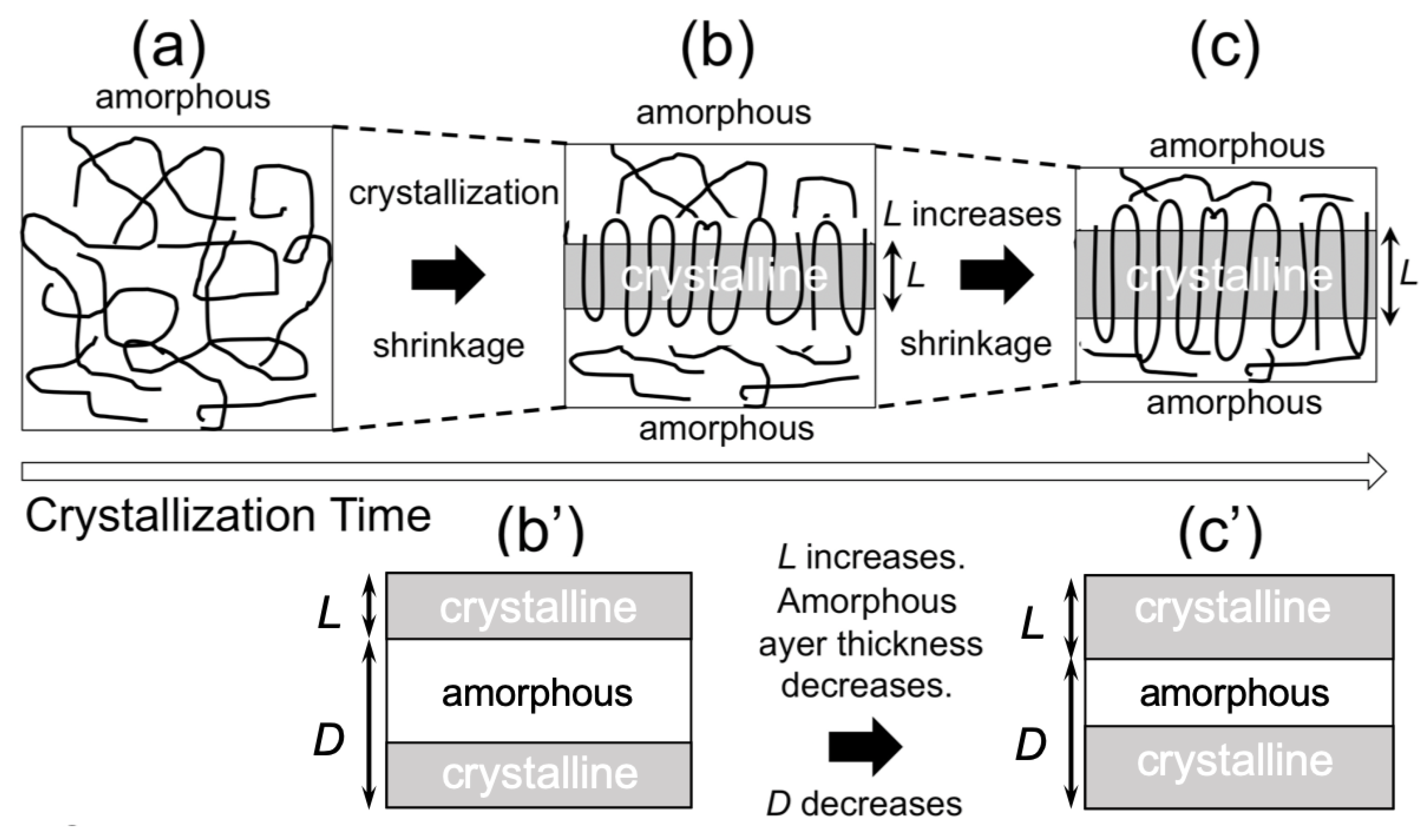
Figure 9b), the average lamellar thickness increases with time, which is reasonable as a crystallization behavior. Therefore, the decreasing behavior of
D is also reasonable, as schematically shown in
Figure 10. Upon crystallization, shrinkage takes place. Since the lamella thickens with time, this results in the decrease of
D (
Figure 10b,c), as an amorphous layer thickness is decreased to a greater extent as compared to the increasing extent of
L (lamellar thickness).
It should be noted here that the average lamellar thickness (
L) did not monotonically increase as a function of time, as the increasing tendency turns over around 7 min for the D0.5 neat specimen or 4 min for the D0.5/SFN(1.0) specimen. The reason why
L decreased a bit before reaching a constant value may be explained by the formation of new lamellae [
17]. It may be explained that in prior to 7 min for the D0.5 specimen or 4 min for the D0.5/SFN(1.0) specimen, a new thin lamellae may be formed from the amorphous region. This argument has been recently reported by our group [
18] for the isothermal crystallization of the D0.5 specimen in the presence of a special plasticizer (OMG) at 100 °C from the melt, where a higher order peak was observed in the Lorentz-corrected SAXS profiles.
Furthermore, as shown in
Figure 9b and
Table 3, the initial average thickness of the lamella (
lc) is thinner for the case of 1% addition of SFN as compared to the neat specimen. This result may explain the mechanism of acceleration of the crystallization by SFN to lower the activation energy of the crystallization with lowering of the thickness of the critical nucleus [
11,
12]. Note here that
L = 6.3 nm is the maximum available for the D1.4 sample because of the D moiety (optical impurity), which is distributed with every 70 repeating units of L moieties when the homogeneous distribution of the D moieties in the main chain of PLLA is assumed. Therefore, the final value of
L in
Table 3 for the D1.4 specimens is somewhat larger. This would imply the random distribution of the D moieties in the main chain of PLLA, further meaning that the Gaussian distribution of the L unit repeating length gives its larger value as compared to the average value which results in
L = 6.3 nm.
The ratio
L/
D seems to be the crystallinity in the stacks of the lamella. To check the increasing tendency of this ratio as a function of time during the isothermal crystallization,
L/
D is plotted as a function of time in
Figure 9c. It is then found that the monotonic increase in
L/
D is then leveled off without the overshooting, as is seen in the plot of
L in
Figure 9b. Moreover, there is observed no effect of the SFN. This is very much contrasted with the crystallization behavior shown in
Figure 2. As shown in
Figure 9c,
L/
D is larger than
ϕWAXS (
Figure 2). The reason why
L/
D is larger than
ϕWAXS is because the lamellar stacks are sparsely dispersed in the matrix of polymer melts, and they do not completely fill the specimen space in the early stage [
20]. Such a situation is illustrated in
Figure 11. Then, the lamellar stacks completely fill the space in the specimen in the late stage, where the
L/
D is almost identical to
ϕWAXS. It is noteworthy that there is no effect of SFN on the behavior of
L/
D, as shown in
Figure 9c for the D1.4 neat and D1.4/SFN(1.0) specimens. At this moment, it is unclear that this result is universal, irrespective of the amount of loading. More detailed study is required. As for the D0.5 neat and D0.5/SFN(1.0) specimens, the SFN even reduces the
L/
D value, which is opposed to the overall behavior shown in
Figure 2. This result may suggest that the SFN enhances the formation of the isolated lamellae without stacking.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}