Synthesis of Poly(3-vinylpyridine)-Block-Polystyrene Diblock Copolymers via Surfactant-Free RAFT Emulsion Polymerization

 ,
,  , , and
, , and
Abstract
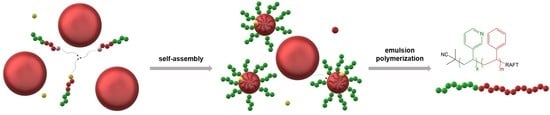
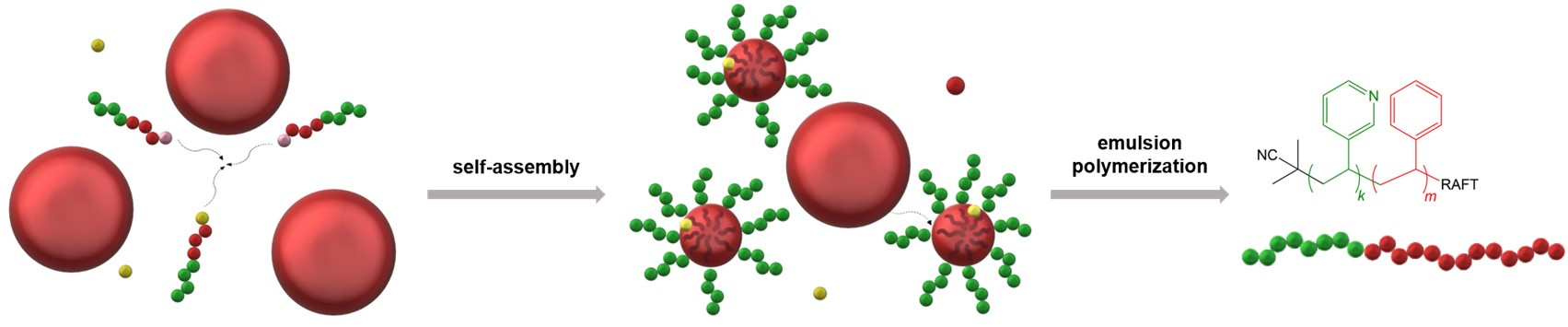
:
1. Introduction
2. Materials and Methods
2.1. Materials
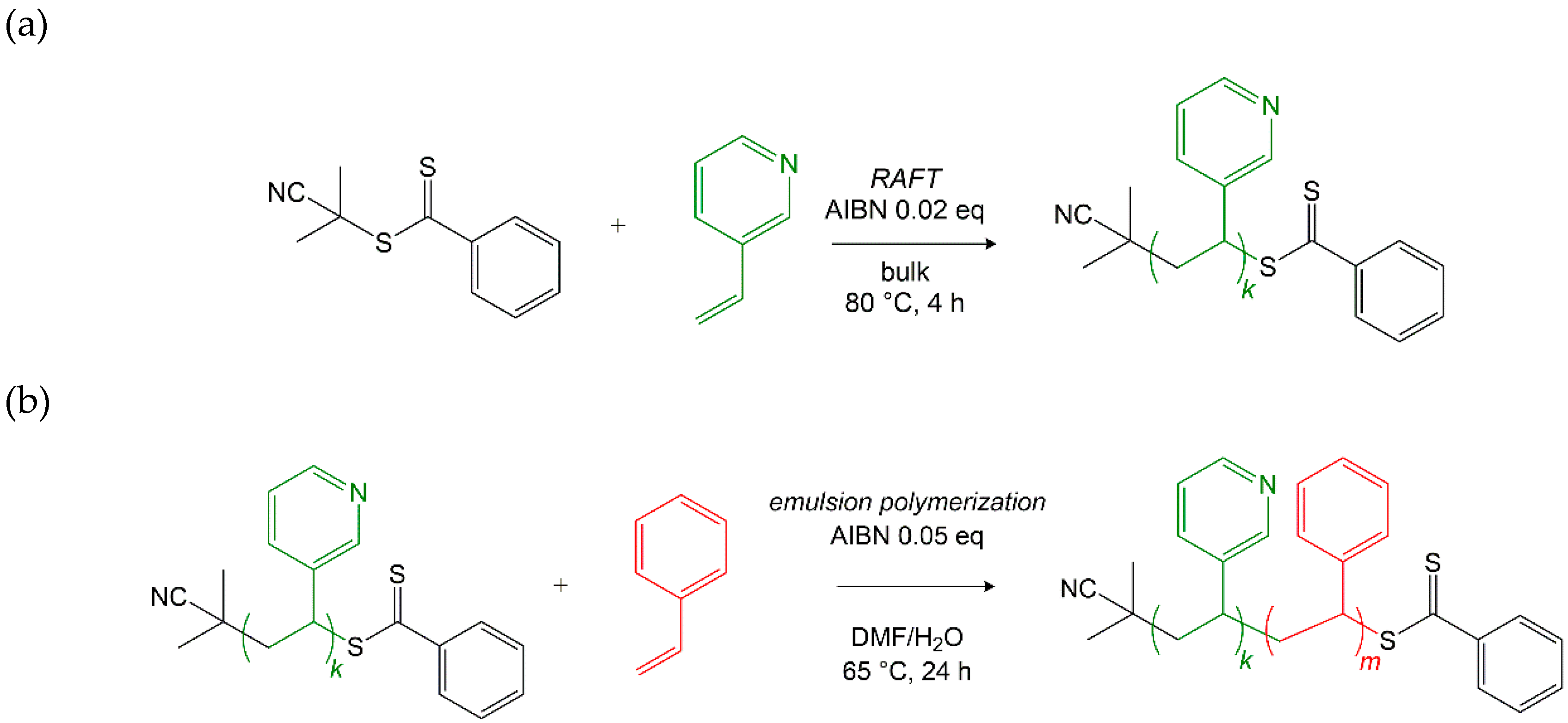
2.1.1. Synthesis of the P3VP macroRAFT Agent
2.1.2. Synthesis of P3VP-b-PS via RAFT Emulsion Polymerization
2.2. Analytics
2.2.1. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.2.2. Size-Exclusion Chromatography (SEC)
2.2.3. Differential Scanning Calorimetry (DSC)
2.2.4. Atomic Force Microscopy (AFM)
2.2.5. Scanning Electron Microscopy (SEM)
2.2.6. Transmission Electron Microscopy (TEM)
3. Results and Discussion
3.1. Preliminary Remarks and Solubility of Aqueous P3VP Solutions
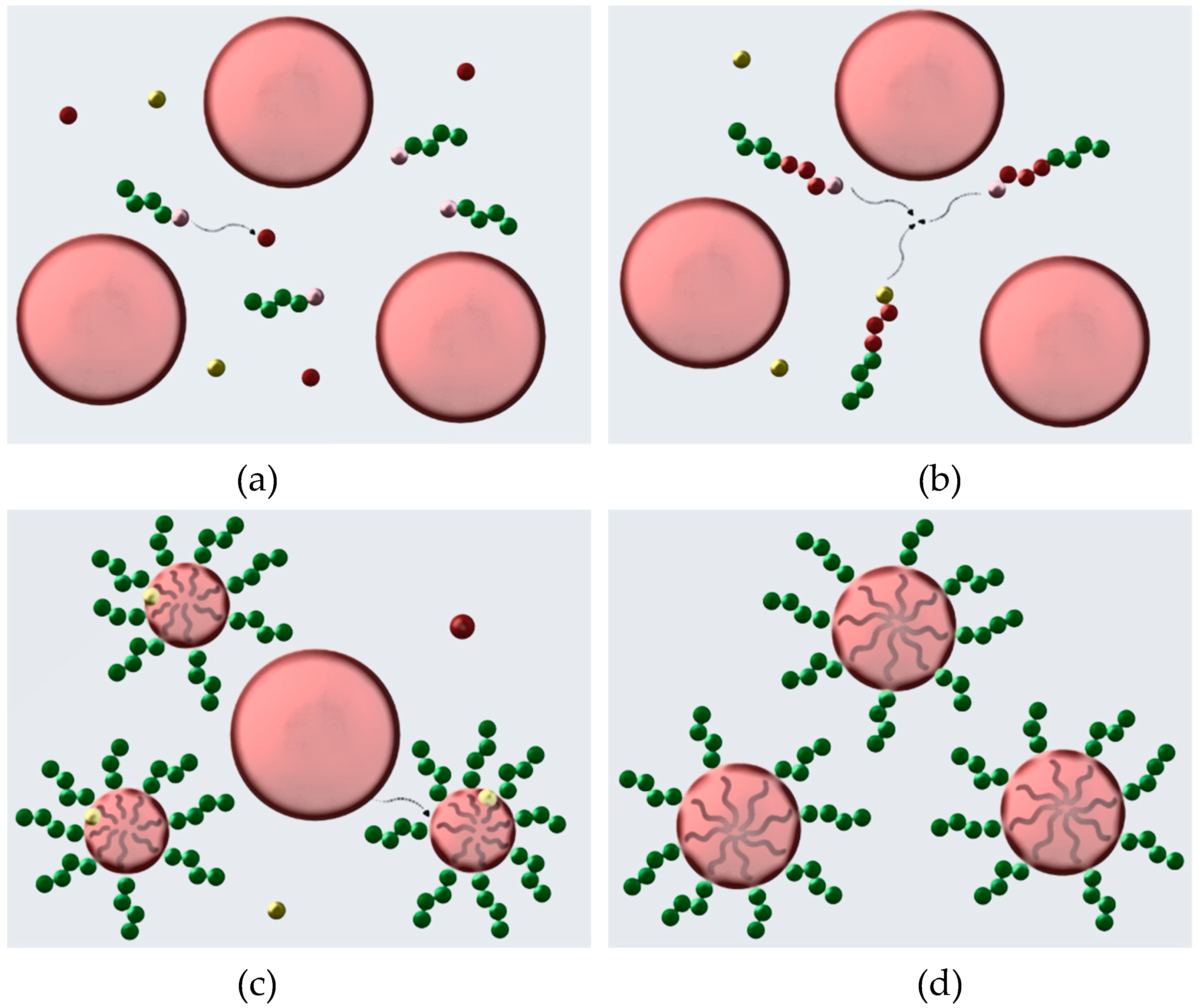
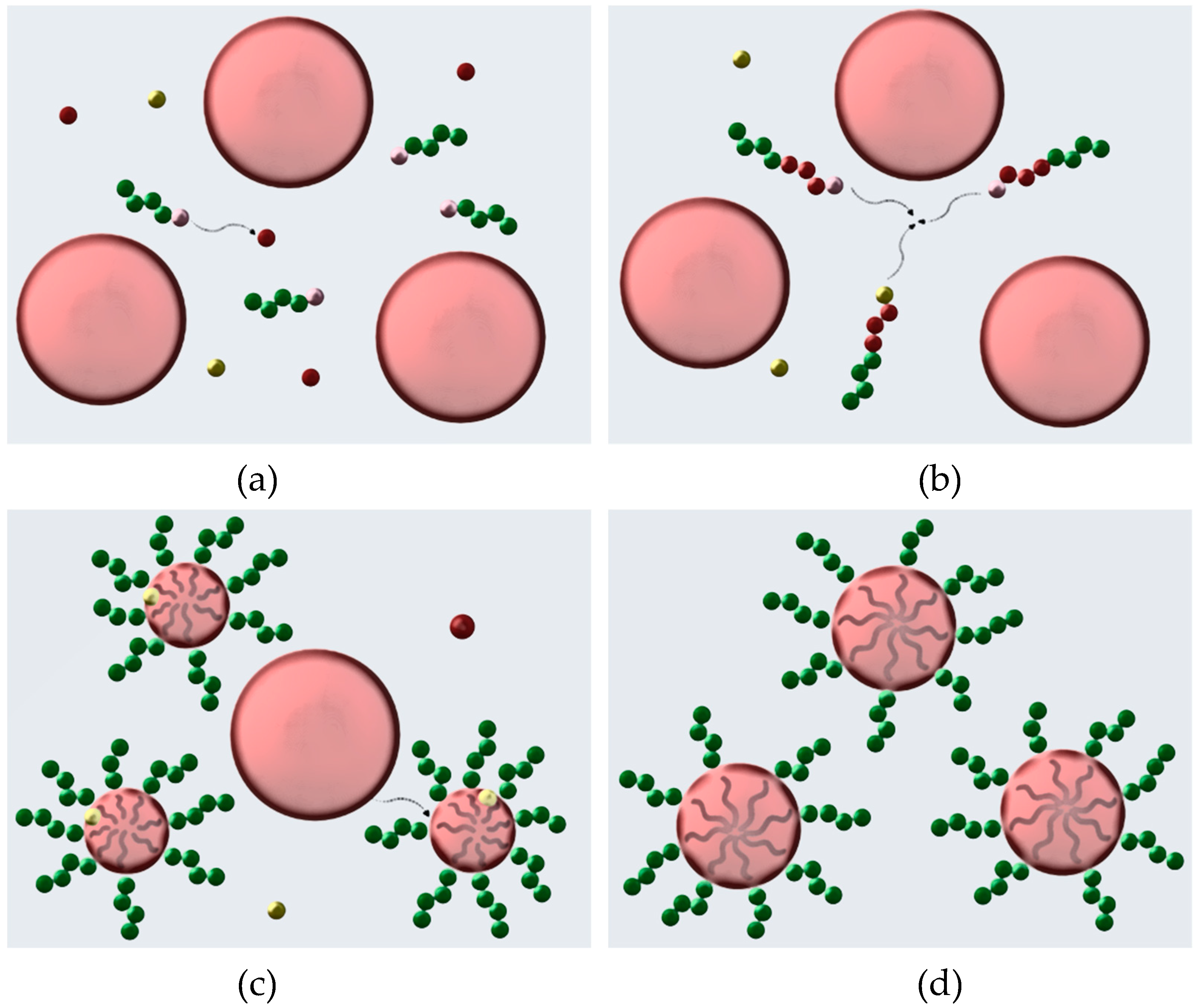
3.2. Mechanism of RAFT Emulsion Polymerization
3.3. Synthesis Results of the P3VP-b-PS Diblock Copolymers
3.4. Thermal and Morphological Characterization of P3VP-b-PS Diblock Copolymers
3.4.1. Thermal Analysis
3.4.2. Morphological Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Sugar-responsive block copolymers by direct RAFT polymerization of unprotected boronic acid monomers. Chem. Commun. 2008, 2477–2479. [Google Scholar] [CrossRef] [PubMed]
- Rizzardo, E.; Solomon, D.H. On the Origins of Nitroxide Mediated Polymerization (NMP) and Reversible Addition–Fragmentation Chain Transfer (RAFT). Aust. J. Chem. 2012, 65, 945–969. [Google Scholar] [CrossRef]
- Ma, J.; Cheng, C.; Sun, G.; Wooley, K.L. Well-defined polymers bearing pendent alkene functionalities via selective RAFT polymerization. Macromolecules 2008, 41, 9080–9089. [Google Scholar] [CrossRef]
- Perrier, S.B. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Pafiti, K.S.; Patrickios, C.S.; Abetz, C.; Abetz, V. High-molecular-weight symmetrical multiblock copolymers: Synthesis by RAFT polymerization and characterization. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4957–4965. [Google Scholar] [CrossRef]
- Rikkou-Kalourkoti, M.; Elladiou, M.; Patrickios, C.S. Synthesis and characterization of hyperbranched amphiphilic block copolymers prepared via self-condensing RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 1310–1319. [Google Scholar] [CrossRef]
- McCormick, C.L.; Lowe, A.B. Aqueous RAFT polymerization: Recent developments in synthesis of functional water-soluble (co) polymers with controlled structures. Acc. Chem. Res. 2004, 37, 312–325. [Google Scholar] [CrossRef]
- Semsarilar, M.; Perrier, S. ‘Green’ reversible addition-fragmentation chain-transfer (RAFT) polymerization. Nat. Chem. 2010, 2, 811–820. [Google Scholar] [CrossRef]
- Luo, J.; Li, M.; Xin, M.; Sun, W. Benzoyl Peroxide/2-Vinylpyridine Synergy in RAFT Polymerization: Synthesis of Poly(2-vinylpyridine) with Low Dispersity at Ambient Temperature. Macromol. Chem. Phys. 2015, 216, 1646–1652. [Google Scholar] [CrossRef]
- Wan, W.-M.; Pan, C.-Y. One-pot synthesis of polymeric nanomaterials via RAFT dispersion polymerization induced self-assembly and re-organization. Polym. Chem. 2010, 1, 1475–1484. [Google Scholar] [CrossRef]
- Zhang, B.Q.; Chen, G.D.; Pan, C.Y.; Luan, B.; Hong, C.Y. Preparation, characterization, and thermal properties of polystyrene-block-quaternized poly (4-vinylpyridine)/Montmorillonite nanocomposites. J. Appl. Polym. Sci. 2006, 102, 1950–1958. [Google Scholar] [CrossRef]
- Luo, J.; Li, M.; Xin, M.; Sun, W.; Xiao, W. Visible Light Induced RAFT Polymerization of 2-Vinylpyridine without Exogenous Initiators or Photocatalysts. Macromol. Chem. Phys. 2016, 217, 1777–1784. [Google Scholar] [CrossRef]
- Arita, T.; Buback, M.; Janssen, O.; Vana, P. RAFT-Polymerization of Styrene up to High Pressure: Rate Enhancement and Improved Control. Macromol. Rapid Commun. 2004, 25, 1376–1381. [Google Scholar] [CrossRef]
- Eggers, S.; Abetz, V. Surfactant-Free RAFT Emulsion Polymerization of Styrene Using Thermoresponsive macroRAFT Agents: Towards Smart Well-Defined Block Copolymers with High Molecular Weights. Polymers 2017, 9, 668. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Grzybowski, B. Self-assembly at all scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef]
- Farrell, R.; Fitzgerald, T.; Borah, D.; Holmes, J.; Morris, M. Chemical interactions and their role in the microphase separation of block copolymer thin films. Int. J. Mol. Sci. 2009, 10, 3671–3712. [Google Scholar] [CrossRef]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A critical appraisal of RAFT-mediated polymerization-induced self-assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef]
- Derry, M.J.; Fielding, L.A.; Armes, S.P. Polymerization-induced self-assembly of block copolymer nanoparticles via RAFT non-aqueous dispersion polymerization. Prog. Polym. Sci. 2016, 52, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Karagoz, B.; Esser, L.; Duong, H.T.; Basuki, J.S.; Boyer, C.; Davis, T.P. Polymerization-Induced Self-Assembly (PISA)—Control over the morphology of nanoparticles for drug delivery applications. Polym. Chem. 2014, 5, 350–355. [Google Scholar] [CrossRef]
- Ferguson, C.J.; Hughes, R.J.; Pham, B.T.; Hawkett, B.S.; Gilbert, R.G.; Serelis, A.K.; Such, C.H. Effective ab initio emulsion polymerization under RAFT control. Macromolecules 2002, 35, 9243–9245. [Google Scholar] [CrossRef]
- Truong, N.P.; Quinn, J.F.; Whittaker, M.R.; Davis, T.P. Polymeric filomicelles and nanoworms: Two decades of synthesis and application. Polym. Chem. 2016, 7, 4295–4312. [Google Scholar] [CrossRef]
- Yeow, J.; Boyer, C. Photoinitiated Polymerization-Induced Self-Assembly (Photo-PISA): New Insights and Opportunities. Adv. Sci. 2017, 4, 1700137. [Google Scholar] [CrossRef] [PubMed]
- Lane, W. Determination of Solubility of Styrene in Water and of Water in Styrene. Ind. Eng. Chem. Anal. Ed. 1946, 18, 295–296. [Google Scholar] [CrossRef]
- Prescott, S.W.; Ballard, M.J.; Rizzardo, E.; Gilbert, R.G. Successful use of RAFT techniques in seeded emulsion polymerization of styrene: Living character, RAFT agent transport, and rate of polymerization. Macromolecules 2002, 35, 5417–5425. [Google Scholar] [CrossRef]
- Mueller, A.H.; Yan, D.; Litvinenko, G.; Zhuang, R.; Dong, H. Kinetic analysis of “living” polymerization processes exhibiting slow equilibria. 2. Molecular weight distribution for degenerative transfer (Direct activity exchange between active and “dormant” species) at constant monomer concentration. Macromolecules 1995, 28, 7335–7338. [Google Scholar] [CrossRef]
- Smulders, W.; Monteiro, M.J. Seeded Emulsion Polymerization of Block Copolymer Core− Shell Nanoparticles with Controlled Particle Size and Molecular Weight Distribution Using Xanthate-Based RAFT Polymerization. Macromolecules 2004, 37, 4474–4483. [Google Scholar] [CrossRef]
- Du, J.; Willcock, H.; Patterson, J.P.; Portman, I.; O’Reilly, R.K. Self-Assembly of Hydrophilic Homopolymers: A Matter of RAFT End Groups. Small 2011, 7, 2070–2080. [Google Scholar] [CrossRef] [Green Version]
- Lang, X.; Patrick, A.D.; Hammouda, B.; Hore, M.J. Chain terminal group leads to distinct thermoresponsive behaviors of linear PNIPAM and polymer analogs. Polymer 2018, 145, 137–147. [Google Scholar] [CrossRef]
- Akpinar, B.; Fielding, L.A.; Cunningham, V.J.; Ning, Y.; Mykhaylyk, O.O.; Fowler, P.W.; Armes, S.P. Determining the effective density and stabilizer layer thickness of sterically stabilized nanoparticles. Macromolecules 2016, 49, 5160–5171. [Google Scholar] [CrossRef]
- Thompson, K.L.; Derry, M.J.; Hatton, F.L.; Armes, S.P. Long-term stability of n-alkane-in-water pickering nanoemulsions: Effect of aqueous solubility of droplet phase on Ostwald ripening. Langmuir 2018, 34, 9289–9297. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.E.; Lee, H.; Choe, S. Synthesis of Functionalized Monodisperse Poly(methyl methacrylate) Nanoparticles by a RAFT Agent Carrying Carboxyl End Group. Macromolecules 2004, 37, 5565–5571. [Google Scholar] [CrossRef]
- Barner-Kowollik, C. Handbook of RAFT Polymerization; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Lansalot, M.; Davis, T.P.; Heuts, J.P. RAFT miniemulsion polymerization: Influence of the structure of the RAFT agent. Macromolecules 2002, 35, 7582–7591. [Google Scholar] [CrossRef]
- Qiu, J.; Gaynor, S.G.; Matyjaszewski, K. Emulsion polymerization of n-butyl methacrylate by reverse atom transfer radical polymerization. Macromolecules 1999, 32, 2872–2875. [Google Scholar] [CrossRef]
- Yamamoto, T. Synthesis of micron-sized polymeric particles in soap-free emulsion polymerization using oil-soluble initiators and electrolytes. Colloid Polym. Sci. 2012, 290, 1023–1031. [Google Scholar] [CrossRef]
- Alberda van Ekenstein, G.; Meyboom, R.; Ten Brinke, G.; Ikkala, O. Determination of the Flory− Huggins Interaction Parameter of Styrene and 4-Vinylpyridine Using Copolymer Blends of Poly (styrene-Co-4-Vinylpyridine) and Polystyrene. Macromolecules 2000, 33, 3752–3756. [Google Scholar] [CrossRef]
- Zha, W.; Han, C.D.; Lee, D.H.; Han, S.H.; Kim, J.K.; Kang, J.H.; Park, C. Origin of the Difference in Order− Disorder Transition Temperature between Polystyrene-block-poly (2-vinylpyridine) and Polystyrene-block-poly (4-vinylpyridine) Copolymers. Macromolecules 2007, 40, 2109–2119. [Google Scholar] [CrossRef]
- Shull, K.R.; Kramer, E.J.; Hadziioannou, G.; Tang, W. Segregation of block copolymers to interfaces between immiscible homopolymers. Macromolecules 1990, 23, 4780–4787. [Google Scholar] [CrossRef]
- Volker Abetz, P.F.W.S. Phase Behaviour and Morphologies of Block Copolymers. In Block Copolymers I; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Lee, D.H.; Cho, H.; Yoo, S.; Park, S. Ordering evolution of block copolymer thin films upon solvent-annealing process. J. Colloid Interface Sci. 2012, 383, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, D.H.; Xu, J.; Kim, B.; Hong, S.W.; Jeong, U.; Xu, T.; Russell, T.P. Macroscopic 10-terabit–per–square-inch arrays from block copolymers with lateral order. Science 2009, 323, 1030–1033. [Google Scholar] [CrossRef]
- Jung, Y.S.; Ross, C.A. Orientation-controlled self-assembled nanolithography using a polystyrene−polydimethylsiloxane block copolymer. Nano Lett. 2007, 7, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Gu, X.; Huh, J.; Xiao, S.; Russell, T.P. Circular nanopatterns over large areas from the self-assembly of block copolymers guided by shallow trenches. ACS Nano 2011, 5, 2855–2860. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Stevens, J.; Villa, J.; Goldbach, J.T.; Guarini, K.W.; Black, C.T.; Hawker, C.J.; Russell, T.P. Block copolymer surface reconstuction: A reversible route to nanoporous films. Adv. Funct. Mater. 2003, 13, 698–702. [Google Scholar] [CrossRef]
- Gowd, E.B.; Nandan, B.; Vyas, M.K.; Bigall, N.C.; Eychmüller, A.; Schlörb, H.; Stamm, M. Highly ordered palladium nanodots and nanowires from switchable block copolymer thin films. Nanotechnology 2009, 20, 415302. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, B.; Wang, J.Y.; Russell, T.P. Fabrication of highly ordered silicon oxide dots and stripes from block copolymer thin films. Adv. Mater. 2008, 20, 681–685. [Google Scholar] [CrossRef]
- Lynd, N.A.; Meuler, A.J.; Hillmyer, M.A. Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 2008, 33, 875–893. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer a | b (kDa) | b (kDa) | c (kDa) | Đ |
|---|---|---|---|---|
| P3VP35-PS6553 | 60 | 68 | 53 | 1.1 |
| P3VP28-PS7268 | 64 | 99 | 68 | 1.5 |
| Polymer | (kDa) | Tg,PS (°C) | Tg,P3VP (°C) |
|---|---|---|---|
| P3VP19 | 19 | NA | 121 |
| P3VP35-PS6553 | 53 | 102 | 122 |
| P3VP28-PS7268 | 68 | 106 | 123 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieswandt, K.; Georgopanos, P.; Abetz, C.; Filiz, V.; Abetz, V. Synthesis of Poly(3-vinylpyridine)-Block-Polystyrene Diblock Copolymers via Surfactant-Free RAFT Emulsion Polymerization. Materials 2019, 12, 3145. https://doi.org/10.3390/ma12193145
Nieswandt K, Georgopanos P, Abetz C, Filiz V, Abetz V. Synthesis of Poly(3-vinylpyridine)-Block-Polystyrene Diblock Copolymers via Surfactant-Free RAFT Emulsion Polymerization. Materials. 2019; 12(19):3145. https://doi.org/10.3390/ma12193145
Chicago/Turabian StyleNieswandt, Katharina, Prokopios Georgopanos, Clarissa Abetz, Volkan Filiz, and Volker Abetz. 2019. "Synthesis of Poly(3-vinylpyridine)-Block-Polystyrene Diblock Copolymers via Surfactant-Free RAFT Emulsion Polymerization" Materials 12, no. 19: 3145. https://doi.org/10.3390/ma12193145