Nanocrystalline Transition-Metal Gallium Oxide Spinels from Acetylacetonate Precursors via Solvothermal Synthesis


1. Introduction
 0.33M2.67O4 with vacant sites ) and migration of the cations to usually unoccupied sites within the close-packed anion array. These possibilities mean that structural disorder may be present that requires careful structural analysis using probes of both long-range and, especially, local-range order. Transition-metal cations are commonly found in oxide spinels, with their specific preferences for octahedral and tetrahedral coordination leading to physical properties that can potentially be tuned by composition. This can lead to, for example, cooperative magnetism, electronic and catalytic properties. The case of ferrites particularly exemplifies the interest, and applications, in magnetism of spinel oxides [2]. Contemporary applications in the field of energy are heavily researched in spinel-structured materials, such as in photocatalysis [3], electrocatalysis [4] and battery cathode materials [5].
0.33M2.67O4 with vacant sites ) and migration of the cations to usually unoccupied sites within the close-packed anion array. These possibilities mean that structural disorder may be present that requires careful structural analysis using probes of both long-range and, especially, local-range order. Transition-metal cations are commonly found in oxide spinels, with their specific preferences for octahedral and tetrahedral coordination leading to physical properties that can potentially be tuned by composition. This can lead to, for example, cooperative magnetism, electronic and catalytic properties. The case of ferrites particularly exemplifies the interest, and applications, in magnetism of spinel oxides [2]. Contemporary applications in the field of energy are heavily researched in spinel-structured materials, such as in photocatalysis [3], electrocatalysis [4] and battery cathode materials [5].2. Experimental Section
2.1. Materials Synthesis
2.2. Materials Characterisation
3. Results and Discussion
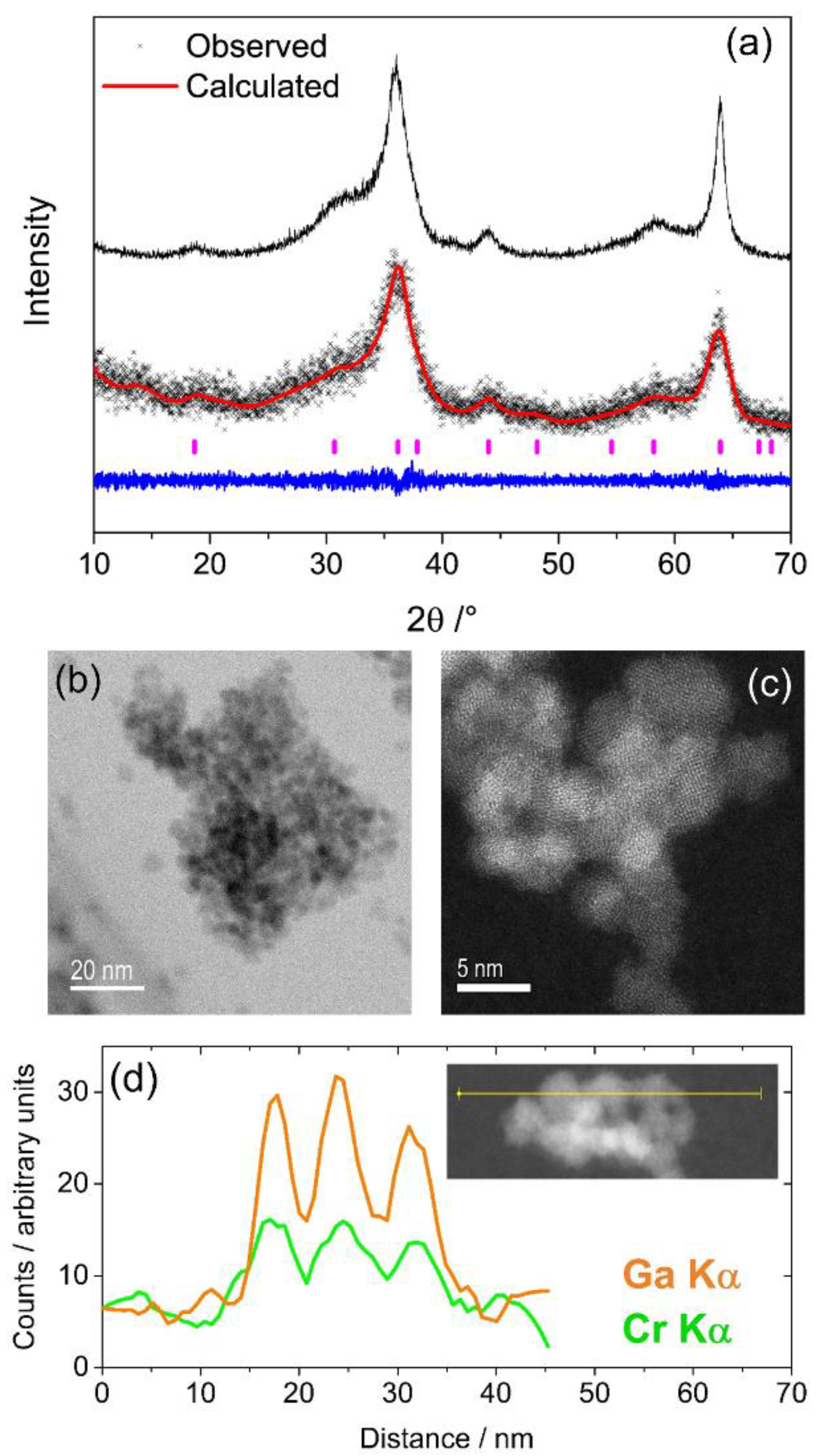
3.1. Cr-Ga2O3
0.33Ga1.867Cr0.8O4, where the vacant sites, , may be octahedral or tetrahedral, or a proportion of each. In our previous work we found that the parent γ-Ga2O3 spinel contained Ga3+ in octahedral and tetrahedral sites, with a distribution that depended on the synthesis method and particle size, but with predominantly octahedral for the most crystalline sample [13]. Since XANES evidence points to the presence of octahedral Cr3+, which is chemically the most likely coordination environment for this cation, we can assume that the mixed Cr-Ga spinel contains a higher proportion of tetrahedral gallium than the binary parent phase. 3.2. Mn-Ga2O3
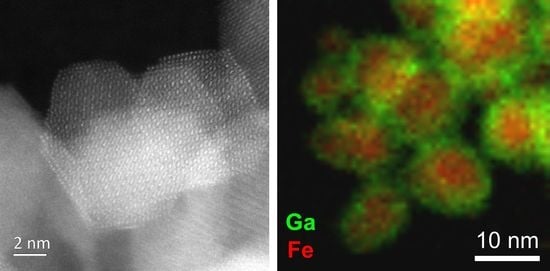
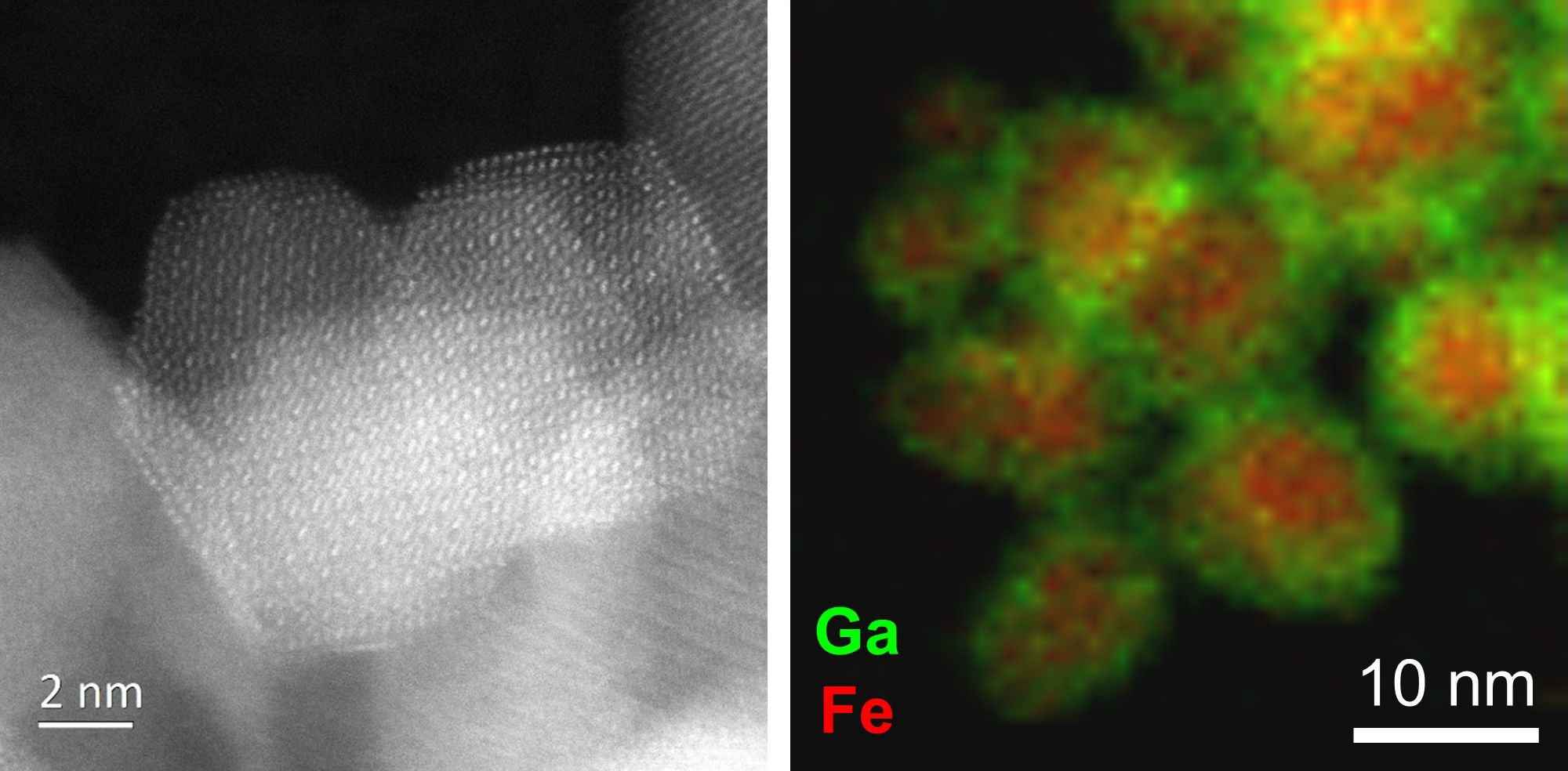
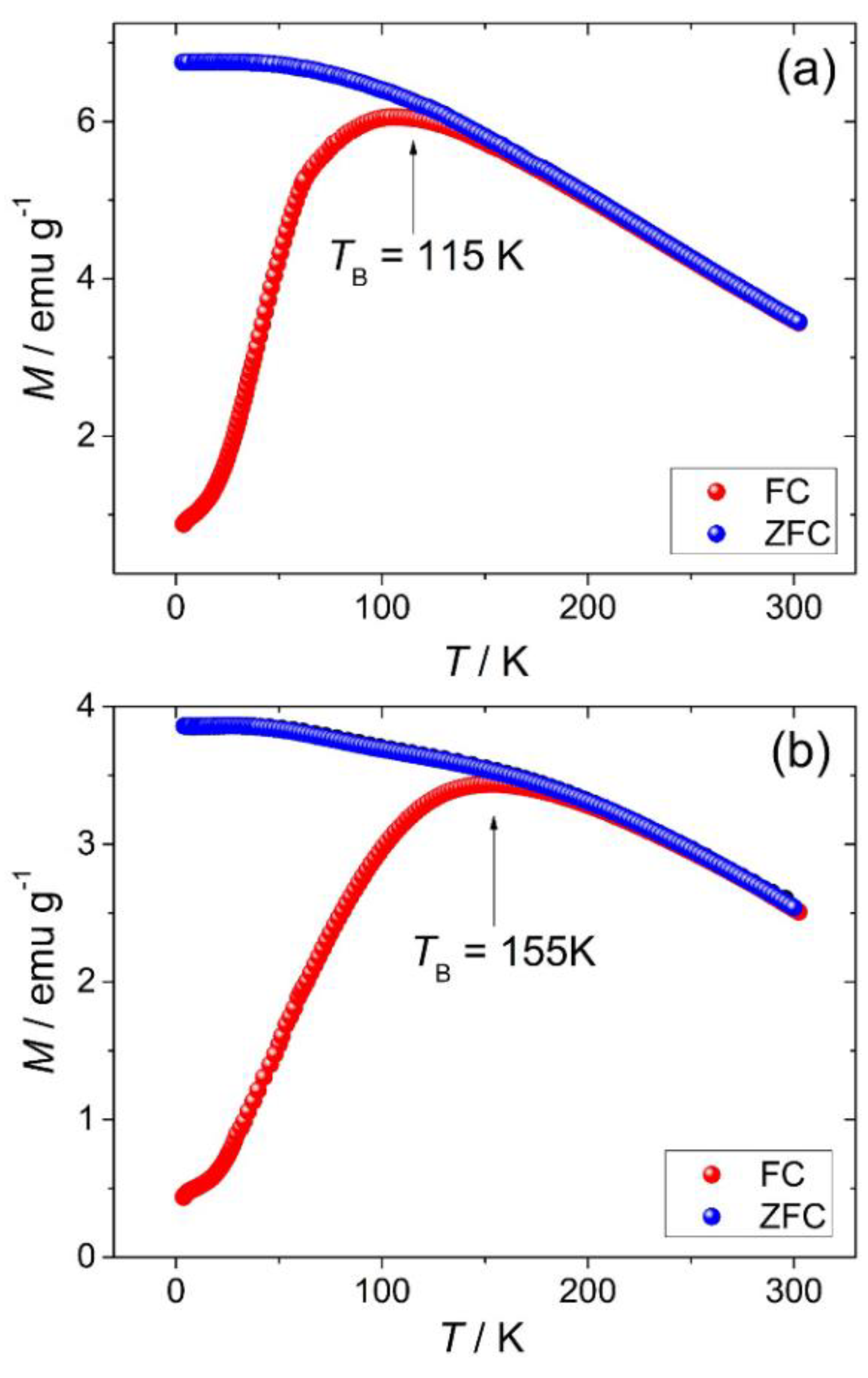
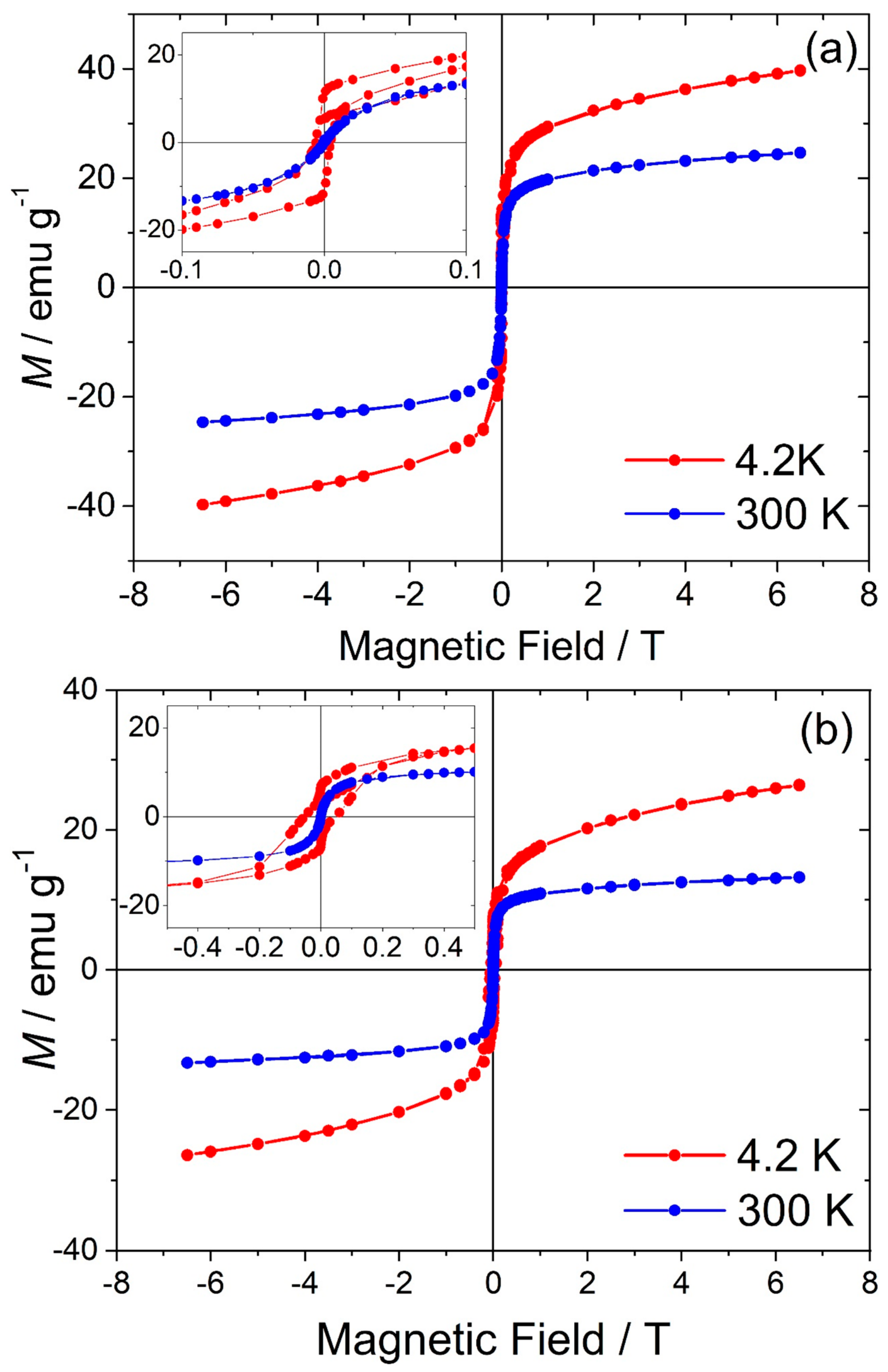
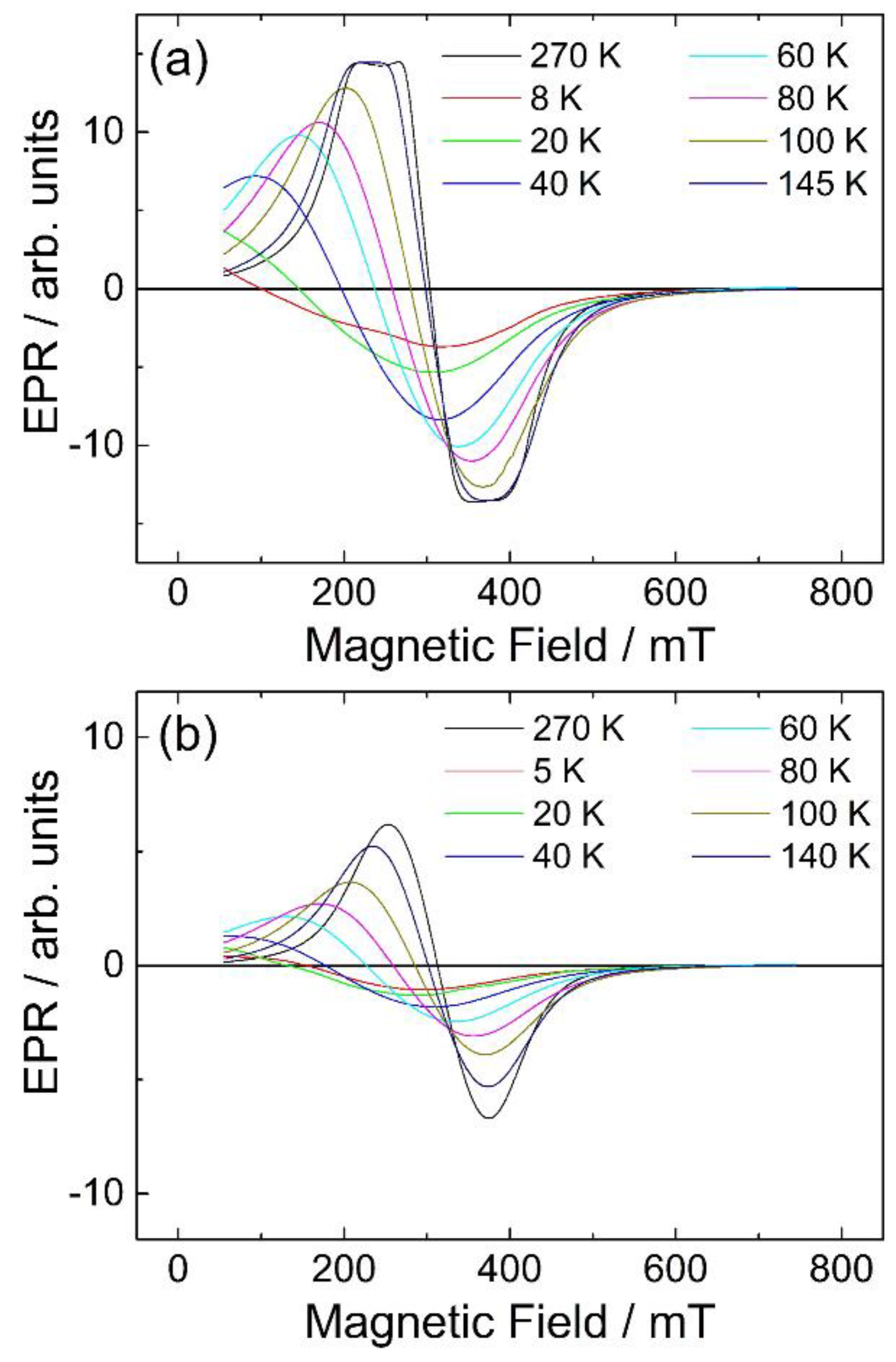
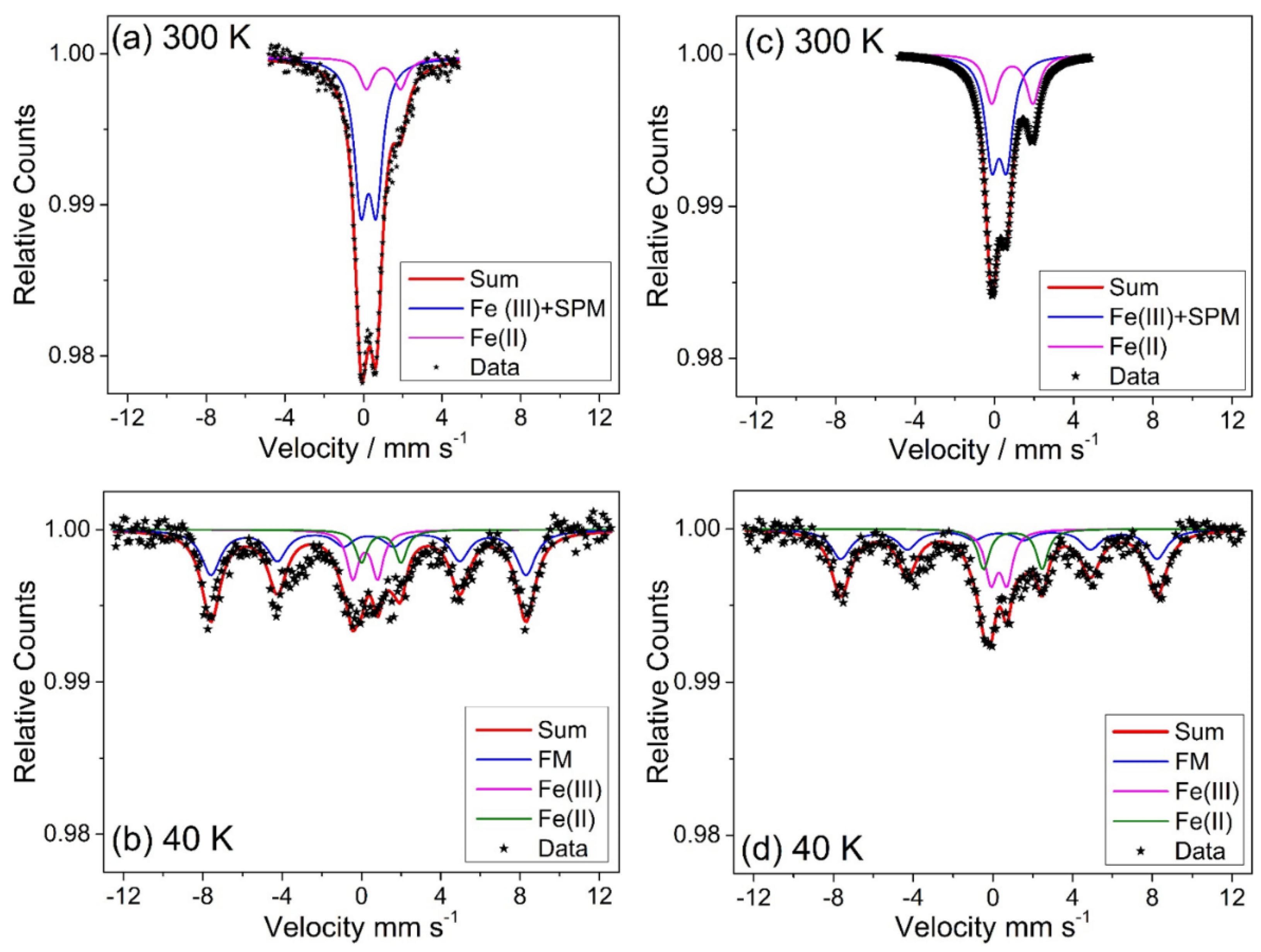
0.18]BO4 has a predicted lattice parameter of 8.36 Å. Additional evidence for the presence of Mn2+ in the new material was provided by thermogravimetric analysis in air, which showed mass gains at around 350 and 500 °C (see Supporting Information), consistent with the work of Laarj et al. on other manganese containing spinels [30], therefore, we conclude that the spinel from solvothermal synthesis contains a mixture of Mn2+ and Mn3+.3.3. Fe-Ga2O3
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sickafus, K.E.; Wills, J.M.; Grimes, N.W. Structure of spinel. J. Am. Ceram. Soc. 1999, 82, 3279–3292. [Google Scholar] [CrossRef]
- Mathew, D.S.; Juang, R.S. An overview of the structure and magnetism of spinel ferrite nanoparticles and their synthesis in microemulsions. Chem. Eng. J. 2007, 129, 51–65. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Bowen, C.; Zhang, P.X.; Li, Z.L.; Yuan, Q.H.; Ren, X.Z.; Deng, L.B. Spinel photocatalysts for environmental remediation, hydrogen generation, CO2 reduction and photoelectrochemical water splitting. J. Mater. Chem. A 2018, 6, 11078–11104. [Google Scholar] [CrossRef]
- Zhao, Q.; Yan, Z.H.; Chen, C.C.; Chen, J. Spinels: Controlled Preparation, Oxygen Reduction/Evolution Reaction Application, and Beyond. Chem. Rev. 2017, 117, 10121–10211. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lee, K.S. Spinel cathodes for advanced lithium ion batteries: A review of challenges and recent progress. Mater. Technol. 2016, 31, 628–641. [Google Scholar] [CrossRef]
- Somiya, S.; Roy, R. Hydrothermal synthesis of fine oxide powders. Bull. Mater. Sci. 2000, 23, 453–460. [Google Scholar] [CrossRef]
- Feng, S.H.; Xu, R.R. New materials in hydrothermal synthesis. Acc. Chem. Res. 2001, 34, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Riman, R.E.; Suchanek, W.L.; Lencka, M.M. Hydrothermal crystallization of ceramics. Ann. Chim. Sci. Mater. 2002, 27, 15–36. [Google Scholar] [CrossRef]
- Walton, R.I. Subcritical solvothermal synthesis of condensed inorganic materials. Chem. Soc. Rev. 2002, 31, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Demazeau, G. Solvothermal reactions: An original route for the synthesis of novel materials. J. Mater. Sci. 2008, 43, 2104–2114. [Google Scholar] [CrossRef]
- Playford, H.Y.; Hannon, A.C.; Barney, E.R.; Walton, R.I. Structures of Uncharacterised Polymorphs of Gallium Oxide from Total Neutron Diffraction. Chem. Eur. J. 2013, 19, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, S.I.; Nikolaev, V.I.; Bougrov, V.E.; Romanov, A.E. Gallium Oxide: Properties and Applications—A Review. Rev. Adv. Mater. Sci. 2016, 44, 63–86. [Google Scholar]
- Playford, H.Y.; Hannon, A.C.; Tucker, M.G.; Dawson, D.M.; Ashbrook, S.E.; Kastiban, R.J.; Sloan, J.; Walton, R.I. Characterization of Structural Disorder in γ-Ga2O3. J. Phys. Chem. C 2014, 118, 16188–16198. [Google Scholar] [CrossRef]
- Kim, S.W.; Iwamoto, S.; Inoue, M. Solvothermal oxidation of gallium metal. Ceram. Intern. 2009, 35, 1603–1609. [Google Scholar] [CrossRef]
- Burnett, D.L.; Harunsani, M.H.; Kashtiban, R.J.; Playford, H.Y.; Sloan, J.; Hannon, A.C.; Walton, R.I. Investigation of some new hydro(solvo)thermal synthesis routes to nanostructured mixed-metal oxides. J. Solid State Chem. 2014, 214, 30–37. [Google Scholar] [CrossRef]
- Cook, D.S.; Wu, Y.; Lienau, K.; More, R.; Kashtiban, R.J.; Magdysyuk, O.V.; Patzke, G.R.; Walton, R.I. Time-Resolved Powder X-ray Diffraction of the Solvothermal Crystallization of Cobalt Gallate Spinel Photocatalyst Reveals Transient Layered Double Hydroxides. Chem. Mater. 2017, 29, 5053–5057. [Google Scholar] [CrossRef]
- Playford, H.Y.; Hannon, A.C.; Tucker, M.G.; Lees, M.R.; Walton, R.I. Total neutron scattering investigation of the structure of a cobalt gallium oxide spinel prepared by solvothermal oxidation of gallium metal. J. Phys. Condens. Matter 2013, 25, 454212. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, M.N.; Chaudhuri, M.K.; Khathing, D.T. Direct Synthesis of “Tris(Acetylacetonato)Manganese(III)”. J. Chem. Soc. Dalton Trans. 1982, 669–670. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C plus. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Dent, A.J.; Cibin, G.; Ramos, S.; Smith, A.D.; Scott, S.M.; Varandas, L.; Pearson, M.R.; Krumpa, N.A.; Jones, C.P.; Robbins, P.E. B18: A core XAS spectroscopy beamline for Diamond. J. Phys. Conf. Ser. 2009, 190, 012039. [Google Scholar] [CrossRef] [Green Version]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Revised Values of Effective Ionic Radii. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 1046–1048. [Google Scholar] [CrossRef]
- Azad, A.K.; Eriksson, S.G.; Yunus, S.M.; Eriksen, J.; Rundlof, H. Synthesis, cation distribution and crystal structure of the spinel type solid solution GaxCoFe1-xCrO4 (0 ≤ x ≤ 1). Phys. B. Condens. Matter 2003, 327, 1–8. [Google Scholar] [CrossRef]
- Boucher, B.; Oles, A. Étude par mesures neutrocristallographiques et magnétiques du spinelle normal antiferromagnétique MnGa2O4. J. Phys. 1966, 27, 51–56. [Google Scholar] [CrossRef]
- Casado, P.G.; Rasines, I. Crystal Data for the Spinels MgGa2O4, MnGa2O4. Z. Kristallogr. 1982, 160, 33–37. [Google Scholar] [CrossRef]
- Boucher, B.; Herpin, A.G.; Oles, A. Antiferromagnetism of the Spinel MnGa2O4. J. Appl. Phys. 1966, 37, 960–961. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, D.S.; Na, C.W.; Park, J. MnGa2O4 and Zn-doped MnGa2O4 1-dimensional nanostructures. J. Phys. Chem. C 2007, 111, 12207–12212. [Google Scholar] [CrossRef]
- Conrad, F.; Bauer, M.; Sheptyakov, D.; Weyeneth, S.; Jaeger, D.; Hametner, K.; Car, P.E.; Patscheider, J.; Gunther, D.; Patzke, G.R. New spinel oxide catalysts for visible-light-driven water oxidation. RSC Adv. 2012, 2, 3076–3082. [Google Scholar] [CrossRef]
- Poix, P. Sur une méthode de détermination des distances cation-oxygène dans les oxydes mixtes à structure spinelle - application des valeurs à quelques cas particuliers. Bull. Soc. Chim. Fr. 1965, 5, 1085–1087. [Google Scholar]
- Laarj, M.; Pignone, I.; El Guendouzi, M.; Tailrades, P.; Rousset, A.; Gillot, B. Quantitative analysis by derivative thermogravimetry of oxidizable cations for finely-divided manganese substituted magnetites. Thermochem. Acta 1989, 152, 187–195. [Google Scholar] [CrossRef]
- Goya, G.F.; Berquo, T.S.; Fonseca, F.C. Static and dynamic magnetic properties of spherical magnetite nanoparticles. J. Appl. Phys. 2003, 94, 3520–3528. [Google Scholar] [CrossRef]
- Liu, C.; Zou, B.; Rondinone, A.J.; Zhang, Z.J. Reverse micelle synthesis and characterization of superparamagnetic MnFe2O4 spinel ferrite nanocrystallites. J. Phys Chem. B 2000, 104, 1141–1145. [Google Scholar] [CrossRef]
- Shipilin, M.A.; Zakharova, I.N.; Shipilin, A.M.; Bachurin, V.I. Mössbauer studies of magnetite nanoparticles. J. Surf. Investig. 2014, 8, 557–561. [Google Scholar] [CrossRef]
- Mitra, S.; Mandal, K. Superparamagnetic behavior in noninteracting NiFe2O4 nanoparticles grown in SiO2 matrix. J. Mater. Manuf. Process. 2007, 22, 444–449. [Google Scholar] [CrossRef]
- Walz, F. The Verwey transition—A topical review. J. Phys. Condens. Matter 2002, 14, R285–R340. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T/K | δ/mm s−1 (± 0.08) | ε/mm s−1 (± 0.15) | BHF/T (± 0.05) | Area (%) (± 1) |
|---|---|---|---|---|---|
| (Fe:Ga) (1:1) | 300 | 0.25 | 0.79 | - | 80 |
| 1.04 | 1.77 | - | 20 | ||
| 40 | 0.35 | 0.00 | 49.4 | 73 | |
| 0.35 | 0.80 | - | 15 | ||
| 0.80 | 2.30 | - | 12 | ||
| (Fe:Ga) (3:7) | 300 | 0.24 | 0.75 | - | 67 |
| 0.91 | 2.07 | - | 33 | ||
| 40 | 0.39 | 0.00 | 49.2 | 66 | |
| 0.30 | 0.78 | - | 25 | ||
| 0.72 | 2.30 | - | 9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cook, D.S.; Kashtiban, R.J.; Krambrock, K.; de Lima, G.M.; Stumpf, H.O.; Lara, L.R.S.; Ardisson, J.D.; Walton, R.I. Nanocrystalline Transition-Metal Gallium Oxide Spinels from Acetylacetonate Precursors via Solvothermal Synthesis. Materials 2019, 12, 838. https://doi.org/10.3390/ma12050838
Cook DS, Kashtiban RJ, Krambrock K, de Lima GM, Stumpf HO, Lara LRS, Ardisson JD, Walton RI. Nanocrystalline Transition-Metal Gallium Oxide Spinels from Acetylacetonate Precursors via Solvothermal Synthesis. Materials. 2019; 12(5):838. https://doi.org/10.3390/ma12050838
Chicago/Turabian StyleCook, Daniel S., Reza J. Kashtiban, Klaus Krambrock, Geraldo M. de Lima, Humberto O. Stumpf, Luciano R. S. Lara, José D. Ardisson, and Richard I. Walton. 2019. "Nanocrystalline Transition-Metal Gallium Oxide Spinels from Acetylacetonate Precursors via Solvothermal Synthesis" Materials 12, no. 5: 838. https://doi.org/10.3390/ma12050838