Unsaturated Poly(Hydroxyalkanoates) for the Production of Nanoparticles and the Effect of Cross-Linking on Nanoparticle Features

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
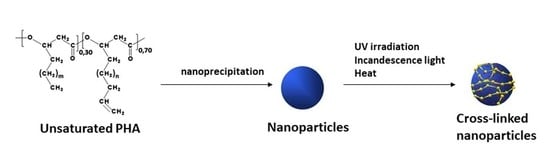
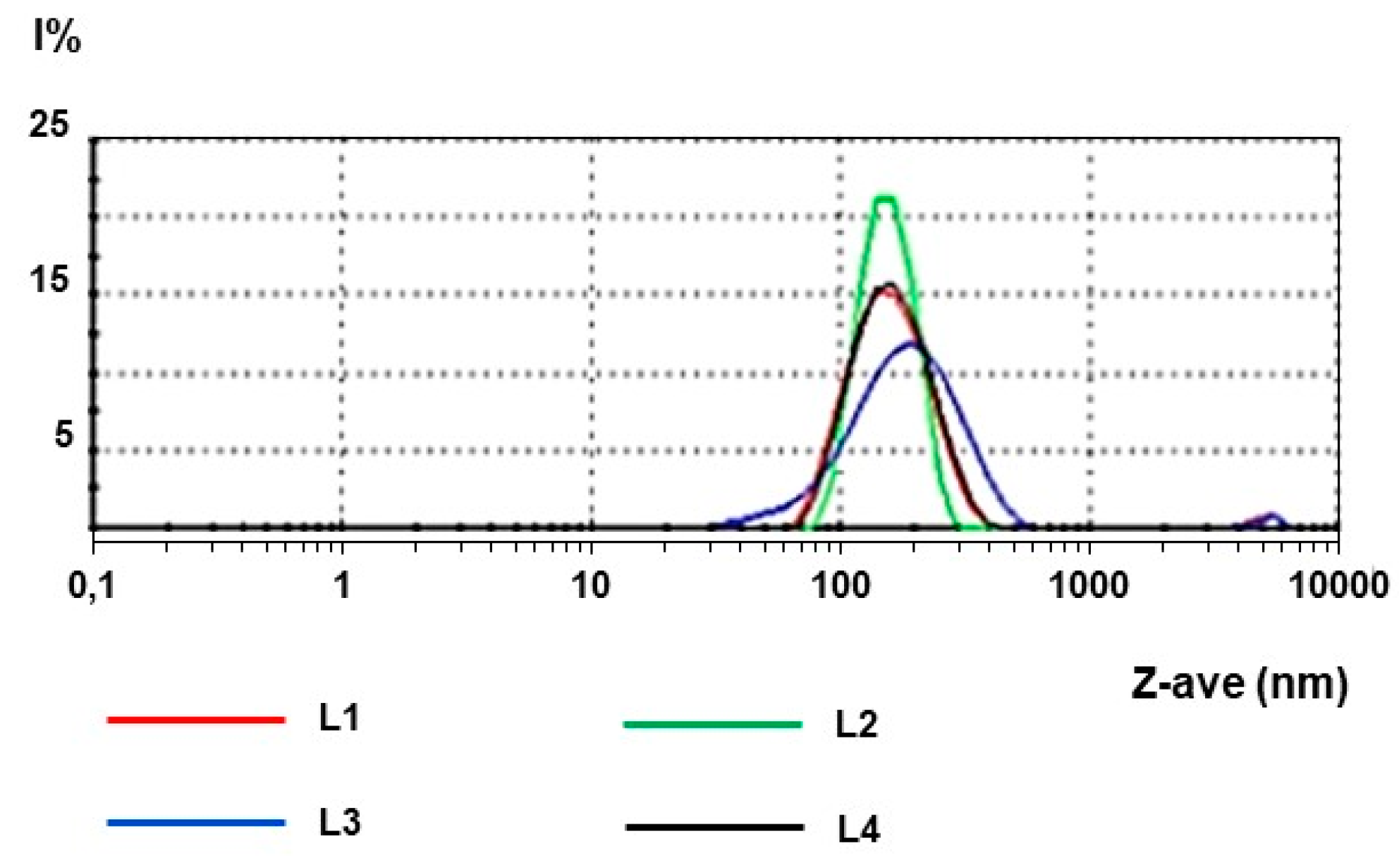
2.1. Physico-Chemical Characterization of Nanoparticles
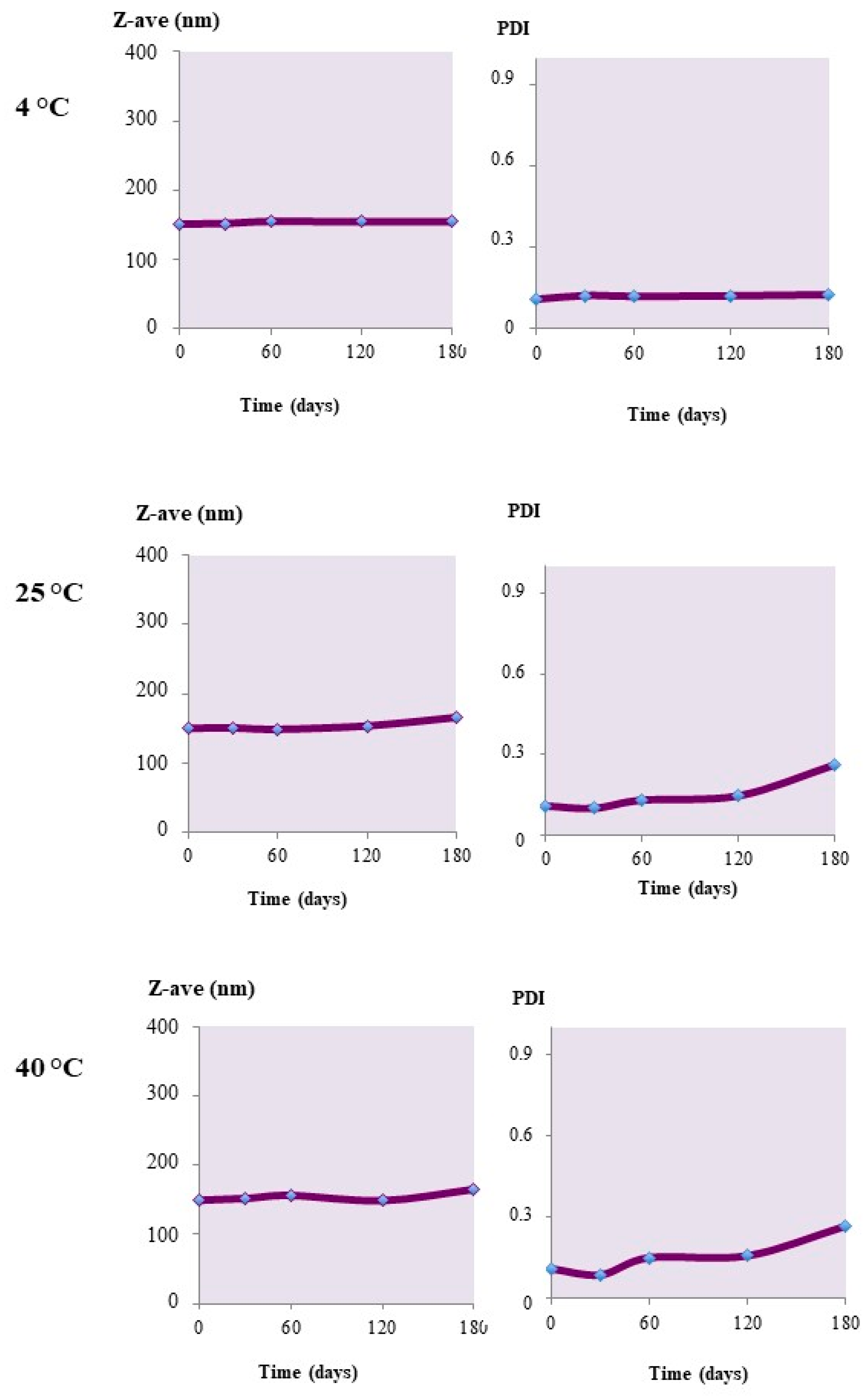
2.2. Stability Studies
2.3. Influence of Cryoprotectants in the Freeze-Drying of Nanoparticles
2.4. Induction of Cross-Linking
2.5. Incandescence Lamp
2.6. UV Irradiation at 300 nm
2.7. Heating
3. Materials and Methods
3.1. Biosynthesis of Poly(3-Hydroxyalkanoate) (Uns-PHA)
3.2. PHA Extraction
3.3. Preparation of Nanoparticles
3.4. Physico-Chemical Characterization of Nanoparticles
3.5. Determination of the Encapsulation Efficiency
3.6. Stability Tests
3.7. Studies on Freeze-Drying and Cryoprotectants
3.8. Cross-Linking Studies
3.8.1. Cross-Linking Induction by a Yellow Light Incandescent Lamp
3.8.2. Cross-Linking Induction by UV Irradiation
3.8.3. Cross-Linking Induction by Heating
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ramachandran, R.; Shanmughavel, P. Preparation and characterization of biopolymeric nanoparticles used in drug delivery. Indian J. Biochem. Biophys. 2010, 47, 56–59. [Google Scholar]
- Shrivastav, A.; Kim, H.-Y.; Kim, Y.-R. Advances in the Applications of Polyhydroxyalkanoate Nanoparticles for Novel Drug Delivery System. BioMed. Res. Int. 2013. [Google Scholar] [CrossRef]
- Lee, J.; Jung, S.G.; Park, C.S.; Kim, H.Y.; Batt, C.A.; Kim, Y.R. Tumor-specific hybrid polyhydroxybutyrate nanoparticle: surface modification of nanoparticle by enzymatically synthesized functional block copolymer. Bioorg. Med. Chem. Lett. 2011, 21, 2941–2944. [Google Scholar] [CrossRef]
- Pignatello, R.; Musumeci, T.; Impallomeni, G.; Carnemolla, G.M.; Puglisi, G.; Ballistreri, A. Poly(3-hydroxybutyrate-co-epsilon-caprolactone) copolymers and poly(3-hydroxybutyrate-co-3-hydroxyvalerate-co-epsilon-caprolactone) terpolymers as novel materials for colloidal drug delivery systems. Eur. J. Pharm. Sci. 2009, 37, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Vilos, C.; Morales, F.A.; Solar, P.A.; Herrera, N.S.; Gonzalez-Nilo, F.D.; Aguayo, D.A.; Mendoza, H.L.; Comer, J.; Bravo, M.L.; Gonzalez, P.A.; et al. Paclitaxel-PHBV nanoparticles and their toxicity to endometrial and primary ovarian cancer cells. Biomaterials 2013, 34, 4098–4108. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Karbasi, S.; Razavi, S.; Zargar, E.N. Poly(hydroxybutyrate)/chitosan aligned electrospun scaffold as a novel substrate for nerve tissue engineering. Adv. Biomed. Res. 2018, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Errico, C.; Bartoli, C.; Chiellini, F.; Chiellini, E. Poly(hydroxyalkanoates)-Based Polymeric Nanoparticles for Drug Delivery. J. Biomed. Biotechnol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.K.; Misra, M.; Hinrichsen, G. Biofibres, biodegradable polymer and composites: An overwiew. Macromol. Mater. Eng. 2000, 276/277, 1–24. [Google Scholar] [CrossRef]
- Steinbuchel, A.; Valentin, H.E. Diversity of bacterial polyhydroxyalkanoic acids. FEMS Microbiol. Lett. 1995, 128, 219–228. [Google Scholar] [CrossRef]
- Zinn, M.; Witholt, B.; Egli, T. Occurrence, synthesis and medical application of bacterial polyhydroxyalkanoate. Adv. Drug Deliv. Rev. 2001, 53, 5–21. [Google Scholar] [CrossRef]
- Rai, R.; Keshavarz, T.; Roether, J.A.; Boccaccini, A.R.; Roy, I. Medium chain length polyhydroxyalkanoates, promising new biomedical materials for the future. Mater. Sci. Eng. R Rep. 2011, 72, 29–47. [Google Scholar] [CrossRef]
- Kwiecień, I.; Radecka, I.; Kwiecień, M.; Adamus, G. Synthesis and structural characterization of bioactive PHA and γ-PGA oligomers for potential applications as a delivery system. Materials 2016, 9, 307. [Google Scholar] [CrossRef]
- Chen, G.Q. Plastics completely syinthesized by bacteria: Polyhydroxyalkanoates. In Plastics from Bacteria; Springer Science & Business Media: Berlin, Germany, 2010. [Google Scholar]
- Kim, Y.B.; Lenz, R.W.; Fuller, R.C. Poly-3-Hydroxyalkanoates containing unsaturated repeating units produced by pseudomonas oleovorans. J. Polym. Sci. Part. A Polym. Chem. 1995, 33, 1367–1374. [Google Scholar] [CrossRef]
- Ashby, R.D.; Foglia, T.A. Poly(hydroxyalkanoate) biosynthesis from triglyceride substrates. Appl. Microbiol. Biotechnol. 1998, 49, 431–437. [Google Scholar] [CrossRef]
- Hazer, B.; Demirel, S.I.; Borcakli, M.; Eroglu, M.S.; Cakmak, M.; Erman, B. Free radical crosslinking of unsaturated bacterial polyesters obtained from soybean oily acids. Polym. Bull. 2001, 46, 389–394. [Google Scholar] [CrossRef]
- Vastano, M.; Pellis, A.; Immirzi, B.; Dal Poggetto, G.; Malinconico, M.; Sannia, G.; Guebitz, G.M.; Pezzella, C. Enzimatic production of clickable and PEGylated recombinant polyhydroxyalkanoates. Green Chem. 2017, 19, 5494–5504. [Google Scholar] [CrossRef]
- Fertier, L.; Koleilat, H.; Stemmelen, M.; Giani, O.; Joly-Duhamel, C.; Lapinte, V.; Robin, J.-J. The use of renewable feedstock in UV-curable materials—A new age for polymers and green chemistry. Prog. Polym. Sci. 2013, 38, 932–962. [Google Scholar] [CrossRef]
- Bassas, M.; Diaz, J.; Rodriguez, E.; Espuny, M.J.; Prieto, M.J.; Manresa, A. Microscopic examination in vivo and in vitro of natural and crosslinked polyunsaturated mcl PHA. Appl. Microbiol. Biotechnol. 2008, 78, 587–596. [Google Scholar] [CrossRef]
- Schmid, M.; Ritter, A.; Grubelnik, A.; Zinn, M. Autoxidation of medium chain length polyhydroxyalkanoate. Biomacromolecules 2007, 8, 579–584. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch | Z-Ave (nm) | PdI | ZP (mV) |
|---|---|---|---|
| L1 | 158.1 | 0.150 | −17.8 |
| L2 | 149.7 | 0.064 | −24.8 |
| L3 | 162.0 | 0.235 | −20.0 |
| L4 | 149.5 | 0.109 | −22.8 |
| Mean | 154.8 | 0.140 | −21.4 |
| ± S.D. | 6.21 | 0.10 | 3.13 |
| Batch | Z-Ave (nm) | PdI | ZP (mV) | EE% |
|---|---|---|---|---|
| L1c | 154.8 | 0.213 | −18.9 | 78.6 |
| L2c | 148.2 | 0.067 | −26.1 | 69.9 |
| L3c | 152.5 | 0.083 | −22.1 | 77.7 |
| L4c | 181.1 | 0.253 | −22.2 | 80.1 |
| Mean | 159.2 | 0.154 | −22.3 | 76.57 |
| ± S.D. | 14.91 | 0.12 | 2.92 | 4.58 |
| Batch | Z-Ave (nm) | PdI | ZP (mV) | EE% |
|---|---|---|---|---|
| L1n | 143.2 | 0.101 | −22.1 | 83.4 |
| L2n | 157.6 | 0.170 | −25.8 | 71.9 |
| L3n | 147.6 | 0.276 | −23.1 | 90.2 |
| L4n | 155.1 | 0.201 | −24.8 | 88.9 |
| Mean | 150.9 | 0.187 | −24.0 | 83.6 |
| ± S.D. | 6.62 | 0.13 | 1.62 | 8.34 |
| Cryoprotectant | Concn. | Z-Ave (nm) | PdI |
|---|---|---|---|
| (% w/v) | (post-lyophilization) | ||
| Trehalose | 5 | 694.9 | 0.878 |
| Trehalose | 10 | 558.6 | 0.531 |
| Lactose | 10 | 623.2 | 0.479 |
| Glucose | 10 | 323.1 | 0.406 |
| Hydroxypropyl-β-cyclodextrin (HP-β-CD) | 1 | 191.4 | 0.184 |
| Nanoparticle Batch | Before | After |
|---|---|---|
| L1 | Soluble | Insoluble |
| L2 | Soluble | Insoluble |
| L3 | Soluble | Insoluble |
| L4 | Soluble | Insoluble |
| Polymer film | Soluble | Insoluble |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignatello, R.; Impallomeni, G.; Cupri, S.; Puzzo, G.; Curcio, C.; Rizzo, M.G.; Guglielmino, S.; Ballistreri, A. Unsaturated Poly(Hydroxyalkanoates) for the Production of Nanoparticles and the Effect of Cross-Linking on Nanoparticle Features. Materials 2019, 12, 868. https://doi.org/10.3390/ma12060868
Pignatello R, Impallomeni G, Cupri S, Puzzo G, Curcio C, Rizzo MG, Guglielmino S, Ballistreri A. Unsaturated Poly(Hydroxyalkanoates) for the Production of Nanoparticles and the Effect of Cross-Linking on Nanoparticle Features. Materials. 2019; 12(6):868. https://doi.org/10.3390/ma12060868
Chicago/Turabian StylePignatello, Rosario, Giuseppe Impallomeni, Sarha Cupri, Giuseppe Puzzo, Claudia Curcio, Maria Giovanna Rizzo, Salvatore Guglielmino, and Alberto Ballistreri. 2019. "Unsaturated Poly(Hydroxyalkanoates) for the Production of Nanoparticles and the Effect of Cross-Linking on Nanoparticle Features" Materials 12, no. 6: 868. https://doi.org/10.3390/ma12060868