The Effect of Heavy Ion Irradiation on the Forward Dissolution Rate of Borosilicate Glasses Studied In Situ and Real Time by Fluid-Cell Raman Spectroscopy

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Glass Synthesis
2.2. Heavy Ion Bombardment
2.3. Raman Spectroscopy
2.4. Experimental Set-Up and Determination of Glass Retreat/Dissolution Rate
3. Results
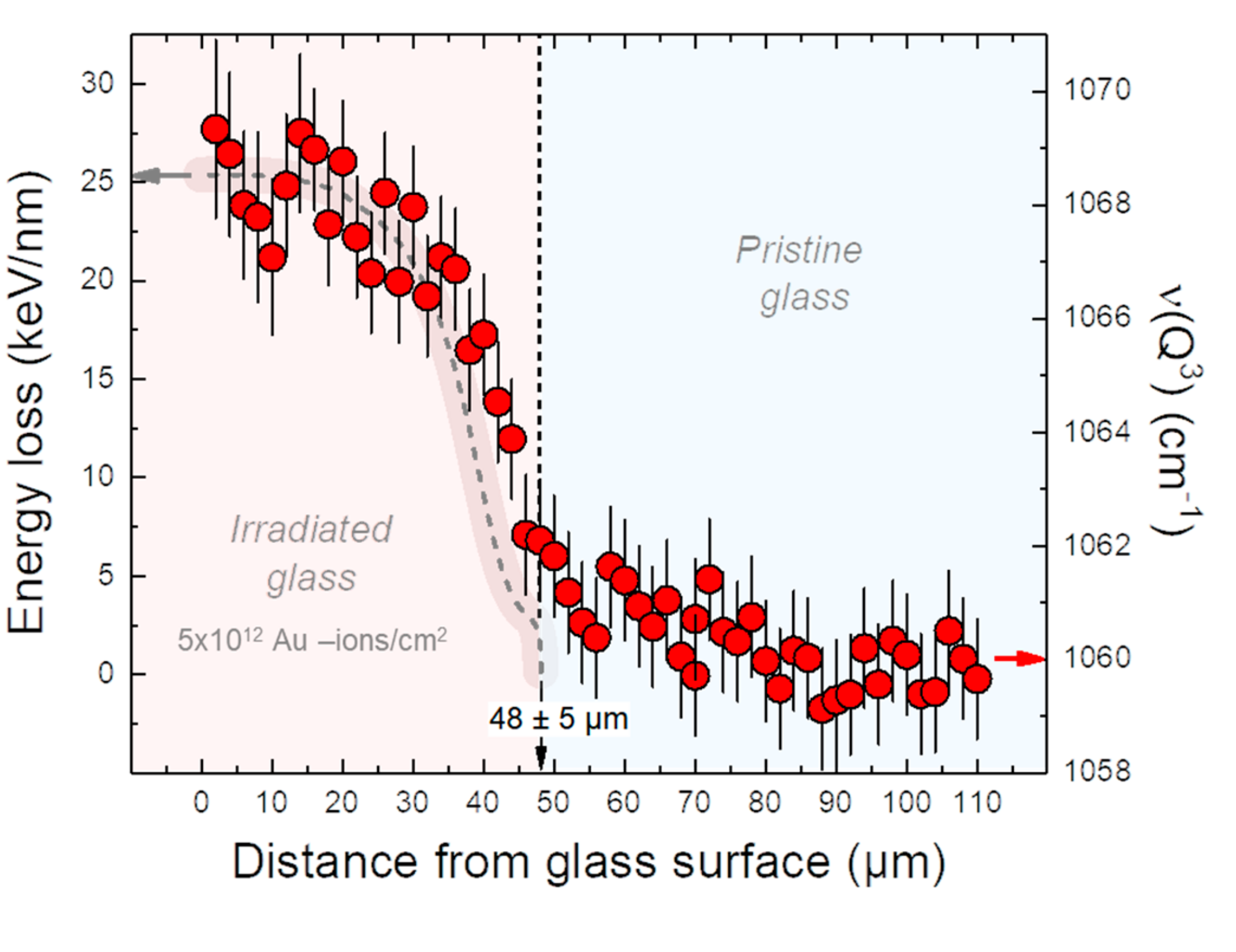
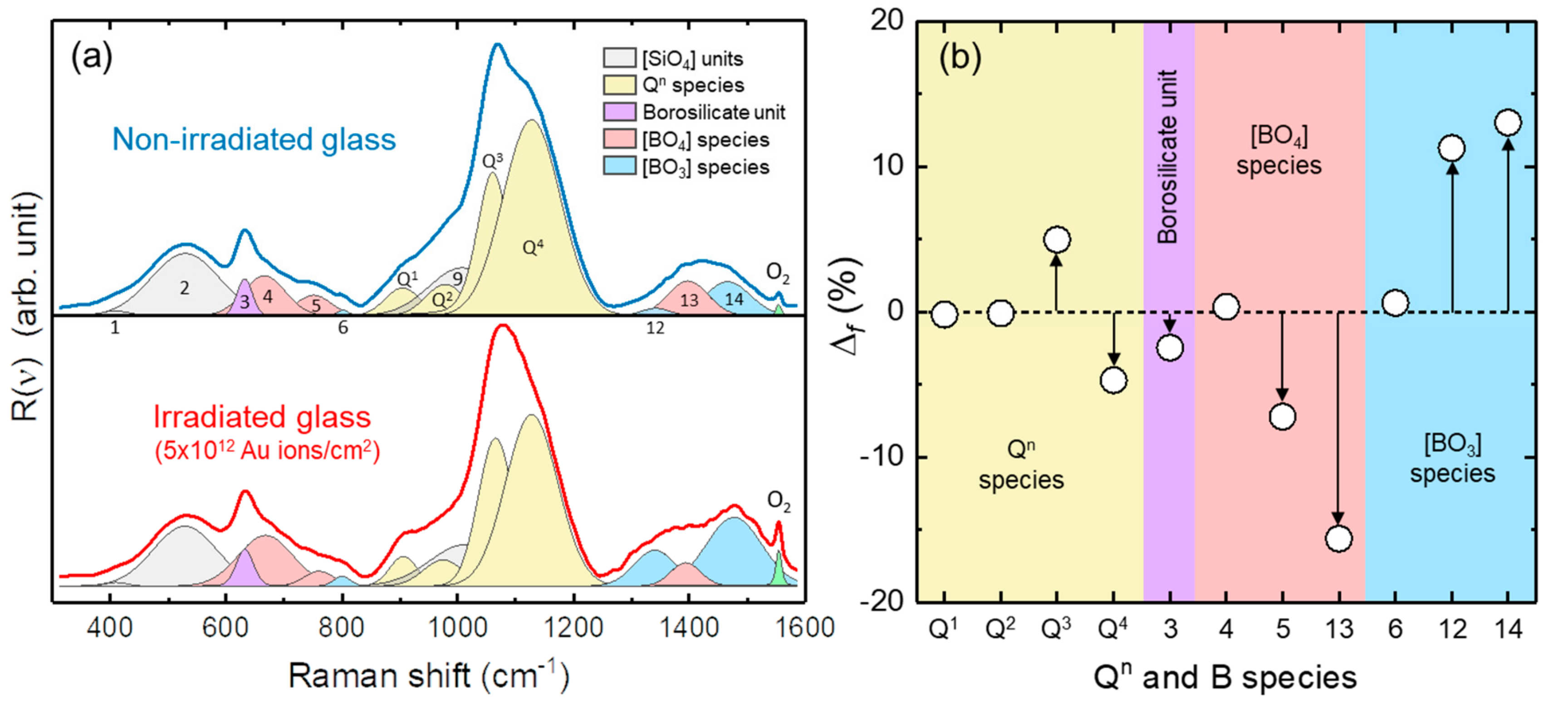
3.1. Structural State of the Irradiated Glass Samples
3.2. Glass Retreat and forward Dissolution Rate
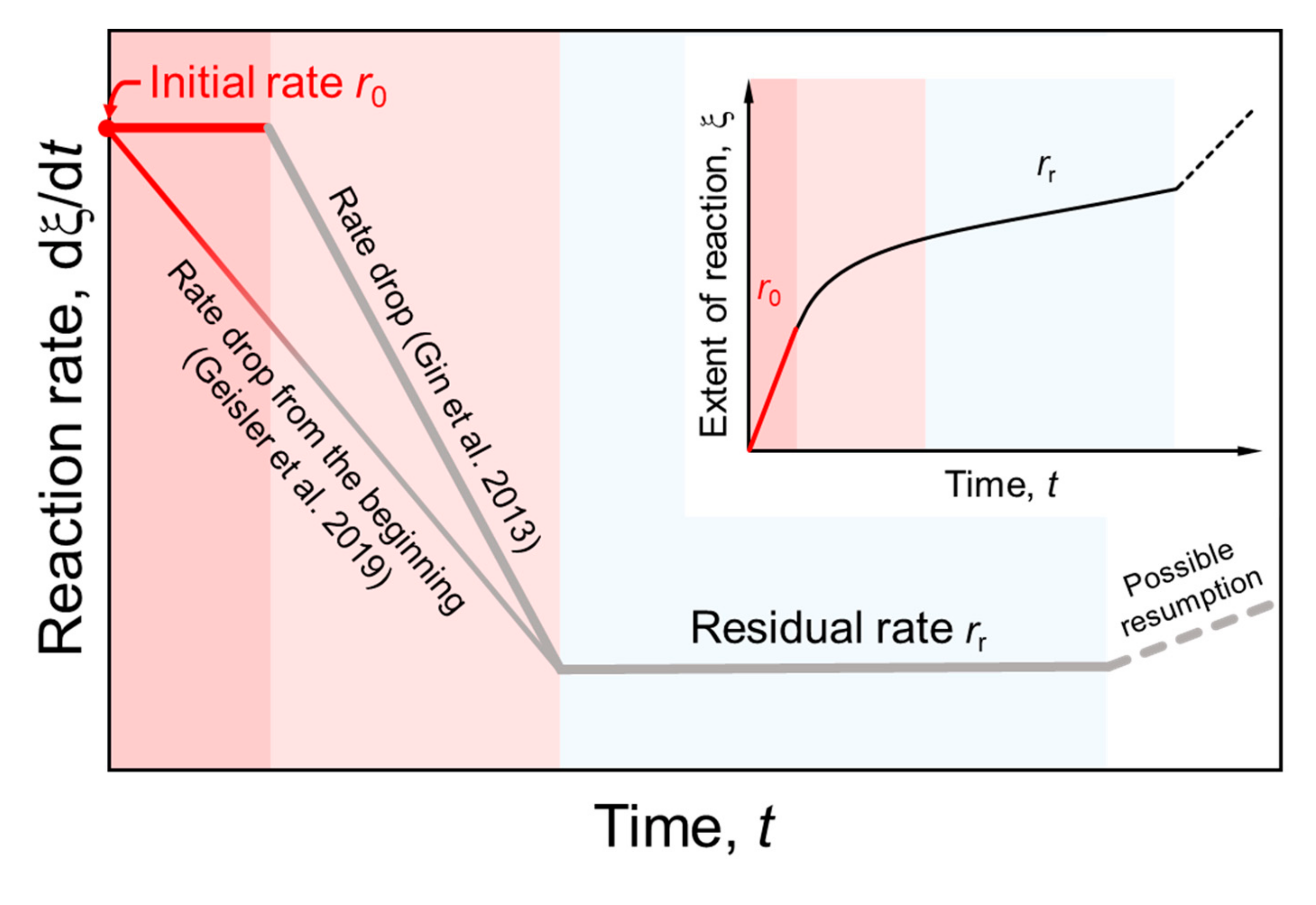
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grambow, B. Nuclear waste glasses—How durable? Elements 2006, 2, 357–364. [Google Scholar] [CrossRef]
- Gin, S.; Abdelouas, A.; Criscenti, L.J.; Ebert, W.L.; Ferrand, K.; Geisler, T.; Harrison, M.T.; Inagaki, Y.; Mitsui, S.; Mueller, K.T.; et al. An international initiative on long-term behavior of high-level nuclear waste glass. Mater. Today 2013, 16, 243–248. [Google Scholar] [CrossRef]
- Icenhower, J.P.; Steefel, C.I. Experimentally determined dissolution kinetics of SON68 glass at 90 °C over a silica saturation interval: Evidence against a linear rate law. J. Nucl. Mater. 2013, 439, 137–147. [Google Scholar] [CrossRef]
- Ojovan, M.I.; Lee, W.E. Glassy wasteforms for nuclear waste immobilization. Metall. Mater. Trans. A 2011, 42, 837–851. [Google Scholar] [CrossRef]
- Grambow, B. A General rate equation for nuclear waste glass corrosion. MRS Proc. 1984, 44, 15–27. [Google Scholar] [CrossRef]
- Hellmann, R.; Cotte, S.; Cadel, E.; Malladi, S.; Karlsson, L.S.; Lozano-Perez, S.; Cabié, M.; Seyeux, A. Nanometre-scale evidence for interfacial dissolution–reprecipitation control of silicate glass corrosion. Nat. Mater. 2015, 14, 307–311. [Google Scholar] [CrossRef]
- Grambow, B.; Müller, R. First-order dissolution rate law and the role of surface layers in glass performance assessment. J. Nucl. Mater. 2001, 298, 112–124. [Google Scholar] [CrossRef]
- Lanford, W.A.; Davis, K.; Lamarche, P.; Laursen, T.; Groleau, R.; Doremus, R.H. Hydration of soda-lime glass. J. Non Cryst. Solids 1979, 33, 249–266. [Google Scholar] [CrossRef]
- Geisler, T.; Janssen, A.; Scheiter, D.; Stephan, T.; Berndt, J.; Putnis, A. Aqueous corrosion of borosilicate glass under acidic conditions: A new corrosion mechanism. J. Non Cryst. Solids 2010, 356, 1458–1465. [Google Scholar] [CrossRef]
- Doremus, R.H. Interdiffusion of hydrogen and alkali ions in a glass surface. J. Non Cryst. Solids 1975, 19, 137–144. [Google Scholar] [CrossRef]
- Abraitis, P.K.; Livens, F.R.; Monteith, J.E.; Small, J.S.; Trivedi, D.P.; Vaughan, D.J.; Wogelius, R.A. The kinetics and mechanisms of simulated british magnox waste glass dissolution as a function of pH, silicic acid activity and time in low temperature aqueous systems. Appl. Geochem. 2000, 15, 1399–1416. [Google Scholar] [CrossRef]
- Geisler, T.; Nagel, T.; Kilburn, M.R.; Janssen, A.; Icenhower, J.P.; Fonseca, R.O.; Grange, M.; Nemchin, A.A. The mechanism of borosilicate glass corrosion revisited. Geochim. Cosmochim. Acta 2015, 158, 112–129. [Google Scholar] [CrossRef]
- Gin, S.; Patrick, J.; Magaly, T.; Sylvain, P.; Schuller, S. Radionuclides containment in nuclear glasses: An overview. Radiochim. Acta 2017, 105, 927–959. [Google Scholar] [CrossRef]
- Lenting, C.; Plümper, O.; Kilburn, M.; Guagliardo, P.; Klinkenberg, M.; Geisler, T. Towards a unifying mechanistic model for silicate glass corrosion. Npj. Mater. Degrad. 2018, 2, 28. [Google Scholar] [CrossRef]
- Poinssot, C.; Gin, S. Long-term behavior science: The cornerstone approach for reliably assessing the long-term performance of nuclear waste. J. Nucl. Mater. 2012, 420, 182–192. [Google Scholar] [CrossRef]
- Vernaz, E.; Gin, S.; Jégou, C.; Ribet, I. Present understanding of R7t7 glass alteration kinetics and their impact on long-term behavior modeling. J. Nucl. Mater. 2001, 298, 27–36. [Google Scholar] [CrossRef]
- Frugier, P.; Gin, S.; Minet, Y.; Chave, T.; Bonin, B.; Godon, N.; Lartigue, J.-E.; Jollivet, P.; Ayral, A.; De Windt, L.; et al. SON68 Nuclear glass dissolution kinetics: Current state of knowledge and basis of the new GRAAL model. J. Nucl. Mater. 2008, 380, 8–21. [Google Scholar] [CrossRef]
- Fournier, M.; Gin, S.; Frugier, P. Resumption of nuclear glass alteration: State of the art. J. Nucl. Mater. 2014, 448, 348–363. [Google Scholar] [CrossRef]
- Ribet, S.; Gin, S. Role of neoformed phases on the mechanisms controlling the resumption of SON68 glass alteration in alkaline media. J. Nucl. Mater. 2004, 324, 152–164. [Google Scholar] [CrossRef]
- Frugier, P.; Fournier, M.; Gin, S. Modeling resumption of glass alteration due to zeolites precipitation. Procedia Earth Planet. Sci. 2017, 17, 340–343. [Google Scholar] [CrossRef]
- Geisler, T.; Dohmen, L.; Lenting, C.; Fritzsche, M.B.K. Real-time in situ observations of reaction and transport phenomena during silicate glass corrosion by fluid-cell Raman spectroscopy. Nat. Mater. 2019, 18, 342–348. [Google Scholar] [CrossRef]
- Advocat, T.; Jollivet, P.; Crovisier, J.L.; Del Nero, M. Long-term alteration mechanisms in water for SON68 radioactive borosilicate glass. J. Nucl. Mater. 2001, 298, 55–62. [Google Scholar] [CrossRef]
- Weber, W.J.; Ewing, R.C.; Angell, C.A.; Arnold, G.W.; Cormack, A.N.; Delaye, J.M.; Griscom, D.L.; Hobbs, L.W.; Navrotsky, A.; Price, D.L.; et al. Radiation effects in glasses used for immobilization of high-level waste and plutonium disposition. J. Mater. Res. 1997, 12, 1948–1978. [Google Scholar] [CrossRef]
- Hobbs, L.W.; Pascucci, M.R. Radiolysis and defect structure in electron-irradiated α-quartz. J. Phys. Colloq. 1980, 41, 237–242. [Google Scholar] [CrossRef]
- Hobbs, L.W. Topology and geometry in the irradiation-induced amorphization of insulators. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1994, 91, 30–42. [Google Scholar] [CrossRef]
- Lang, M.; Tracy, C.L.; Palomares, R.; Zhang, F.; Severin, D.; Bender, M.; Trautmann, C.; Park, C.; Prakapenka, V.B.; Skuratov, V.A.; et al. Characterization of ion-induced radiation effects in nuclear materials using synchrotron x-ray techniques. J. Mater. Res. 2015, 30, 1366–1379. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Ziegler, M.D.; Biersack, J.P. SRIM—The stopping and range of ions in matter (2010). Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2010, 268, 1818–1823. [Google Scholar] [CrossRef]
- Efimov, A. Vibrational spectra, related properties, and structure of inorganic glasses. J. Non Cryst. Solids 1999, 253, 95–118. [Google Scholar] [CrossRef]
- Mysen, B.O.; Finger, L.W.; Virgo, D.; Seifert, F.A. Curve-fitting of Raman spectra of silicate glasses. Am. Mineral. 1982, 67, 686–695. [Google Scholar]
- Mckeown, D.; Muller, I.; Buechele, A.; Pegg, I.L.; Kendziora, C. Structural characterization of high-zirconia borosilicate glasses using Raman spectroscopy. J. Non Cryst. Solids 2000, 262, 126–134. [Google Scholar] [CrossRef]
- Manara, D.; Grandjean, A.; Neuville, D.R. Advances in understanding the structure of borosilicate glasses: A Raman spectroscopy study. Am. Mineral. 2009, 94, 777–784. [Google Scholar] [CrossRef]
- Fukumi, K.; Hayakawa, J.; Komiyama, T. Intensity of Raman band in silicate glasses. J. Non Cryst. Solids 1990, 119, 297–302. [Google Scholar] [CrossRef]
- Neuville, D. Viscosity, structure and mixing in (Ca, Na) silicate melts. Chem. Geol. 2006, 229, 28–41. [Google Scholar] [CrossRef]
- Frantza, J.D.; Mysen, B.O. Raman spectra and structure of BaO-SiO2 SrO-SiO2 and CaO-SiO2 melts to 1600 °C. Chem. Geol. 1995, 121, 155–176. [Google Scholar] [CrossRef]
- Mir, A.H.; Monnet, I.; Boizot, B.; Jégou, C.; Peuget, S. Electron and electron-ion sequential irradiation of borosilicate glasses: Impact of the pre-existing defects. J. Nucl. Mater. 2017, 489, 91–98. [Google Scholar] [CrossRef]
- Zapol, P.; He, H.; Kwon, K.; Criscenti, L. First-Principles Study of Hydrolysis Reaction Barriers in a Sodium Borosilicate Glass. Int. J. Appl. Glass Sci. 2013, 4, 395–407. [Google Scholar] [CrossRef]
- Peuget, S.; Tribet, M.; Mougnaud, S.; Miro, S.; Jégou, C. Radiations effects in ISG glass: from structural changes to long-term aqueous behavior. Npj. Mater. Degrad. 2018, 2, 23. [Google Scholar] [CrossRef]
- Burns, W.G.; Hughes, A.E.; Marples, J.A.C.; Nelson, R.S.; Stoneham, A.M. Effects of radiation on the leach rates of vitrified radioactive waste. J. Nucl. Mater. 1982, 107, 245–270. [Google Scholar] [CrossRef]
- Weber, W.J.; McVay, G.L.; Wald, J.W. Effects of alpha-radiolysis on leaching of a nuclear waste glass. J. Am. Ceram. Soc. 1985, 68, 253–255. [Google Scholar] [CrossRef]
- Peuget, S.; Broudic, V.; Jégou, C.; Frugier, P.; Roudil, D.; Deschanels, X.; Rabiller, H.; Y. Noel, P. Effect of alpha radiation on the leaching behaviour of nuclear glass. J. Nucl. Mater. 2007, 362, 474–479. [Google Scholar] [CrossRef]
- Wellman, D.; Icenhower, J.; Weber, W. Elemental Dissolution Study of Pu-Bearing Borosilicate Glasses. J. Nucl. Mater. 2005, 340, 149–162. [Google Scholar] [CrossRef]
- Marples, J.A.C. Dose rate effects in radiation damage to vitrified radioactive waste. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1988, 32, 480–486. [Google Scholar] [CrossRef]
- Delaye, J.-M.; Kerrache, A.; Gin, S. Topography of borosilicate glass reacting interface under aqueous corrosion. Chem. Phys. Lett. 2013, 588, 180–183. [Google Scholar] [CrossRef]
- Dewan, L.; Hobbs, L.W.; Delaye, J.-M. Topological Analysis of the Structure of Self-irradiated Sodium Borosilicate Glass. J. Non Cryst. Solids 2012, 358, 3427–3432. [Google Scholar] [CrossRef]
- Peuget, S.; Maugeri, E.A.; Charpentier, T.; Mendoza, C.; Moskura, M.; Fares, T.; Bouty, O.; Jégou, C. Comparison of radiation and quenching rate effects on the structure of a sodium borosilicate glass. J. Non Cryst. Solids 2013, 378, 201–212. [Google Scholar] [CrossRef]
- Karakurt, G. Effect of alpha radiation on the physical and chemical properties of silicate glasses. Ph.D. Thesis, Ecole des Mines de Nantes, Nantes, France, 2014. [Google Scholar]
- Mir, A.H.; Peuget, S.; Toulemonde, M.; Bulot, P.; Jégou, C.; Miro, S.; Bouffard, S. Defect recovery and damage reduction in borosilicate glasses under double ion beam irradiation. EPL Europhys. Lett. 2015, 112, 36002. [Google Scholar] [CrossRef]
- Kluth, P.; Schnohr, C.S.; Pakarinen, O.H.; Djurabekova, F.; Sprouster, D.J.; Giulian, R.; Ridgway, M.C.; Byrne, A.P.; Trautmann, C.; Cookson, D.J.; et al. Fine structure in swift heavy ion tracks in amorphous SiO2. Phys. Rev. Lett. 2008, 101, 175503. [Google Scholar] [CrossRef]
- Mir, A.H.; Monnet, I.; Toulemonde, M.; Bouffard, S.; Jégou, C.; Peuget, S. Mono and sequential ion irradiation induced damage formation and damage recovery in oxide glasses: Stopping power dependence of the mechanical properties. J. Nucl. Mater. 2015, 469, 244–250. [Google Scholar] [CrossRef]
- Charpentier, T.; Martel, L.; Mir, A.H.; Somers, J.; Jégou, C.; Peuget, S. Self-healing capacity of nuclear glass observed by NMR spectroscopy. Sci. Rep. 2016, 6, 25499. [Google Scholar] [CrossRef] [PubMed]
- Arnold, G.W. Alkali depletion and ion beam mixing in glasses. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1984, 1, 516–520. [Google Scholar] [CrossRef]
- Stroes-Gascoyne, S.; King, F.; Betteridge, J.S.; Garisto, F. The effects of alpha-radiolysis on UO2 dissolution determined from electrochemical experiments with 238Pu-doped UO2. Radiochim. Acta 2002, 90, 9–11. [Google Scholar] [CrossRef]
- McVay, G.L.; Weber, W.J.; Pederson, L.R. Effects of radiation on the leaching behavior of nuclear waste forms. Nucl. Chem. Waste Manag. 1981, 2, 103–108. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Non-Irradiated TBG | Irradiated TBG | |||
|---|---|---|---|---|---|
| T (°C) r0irr (µm/h) r0 (µm/h) | 85.2 ± 0.2 - 1.61 ± 0.04 | 81.7± 0.1 - 1.00 ± 0.03 | 85.1 ± 0.7 - 2.10 ± 0.03 | 80.0 ± 0.2 5.1 ± 0.4 1.8 ± 0.2 | 82.6 ± 0.3 5.5 ± 0.6 1.5 ± 0.1 |
| Reference | [21] | This study | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lönartz, M.I.; Dohmen, L.; Lenting, C.; Trautmann, C.; Lang, M.; Geisler, T. The Effect of Heavy Ion Irradiation on the Forward Dissolution Rate of Borosilicate Glasses Studied In Situ and Real Time by Fluid-Cell Raman Spectroscopy. Materials 2019, 12, 1480. https://doi.org/10.3390/ma12091480
Lönartz MI, Dohmen L, Lenting C, Trautmann C, Lang M, Geisler T. The Effect of Heavy Ion Irradiation on the Forward Dissolution Rate of Borosilicate Glasses Studied In Situ and Real Time by Fluid-Cell Raman Spectroscopy. Materials. 2019; 12(9):1480. https://doi.org/10.3390/ma12091480
Chicago/Turabian StyleLönartz, Mara Iris, Lars Dohmen, Christoph Lenting, Christina Trautmann, Maik Lang, and Thorsten Geisler. 2019. "The Effect of Heavy Ion Irradiation on the Forward Dissolution Rate of Borosilicate Glasses Studied In Situ and Real Time by Fluid-Cell Raman Spectroscopy" Materials 12, no. 9: 1480. https://doi.org/10.3390/ma12091480