
A DFT/TD-DFT Study on the ESIPT-Type Flavonoid Derivatives with High Emission Intensity


College of Science, Northeast Forestry University, Harbin 150040, China
*
Authors to whom correspondence should be addressed.
Materials 2022, 15(8), 2896; https://doi.org/10.3390/ma15082896
Submission received: 22 March 2022
/
Revised: 6 April 2022
/
Accepted: 11 April 2022
/
Published: 15 April 2022
(This article belongs to the Section Optical and Photonic Materials)
Abstract
:To reveal the influence of different substituents on the excited-state intramolecular proton transfer (ESIPT) process and photophysical properties of 4′-N, N-dimethylamino-3-hydroxyflavone (DMA3HF), two novel molecules (DMA3HF-CN and DMA3HF-NH2) were designed by introducing the classical electron-withdrawing group cyano (-CN) and electron-donating group amino (-NH2). The three molecules in the acetonitrile phase were systematically researched by applying the density functional theory (DFT) and time-dependent DFT (TD-DFT) methods. The excited-state hydrogen bond enhancement mechanism was confirmed, and the hydrogen bond intensity followed the decreasing order of DMA3HF-NH2 > DMA3HF > DMA3HF-CN, which can be explained at the electronic level by natural bond orbital, fuzzy bond order, and frontier molecular orbital analyses. Moreover, we found from the electronic spectra that the fluorescence intensity of the three molecules in keto form is relatively strong. Moreover, the calculated absorption properties indicated that introducing the electron-withdrawing group -CN could significantly improve the absorption of DMA3HF in the ultraviolet band. In summary, the introduction of an electron-donating group -NH2 can promote the ESIPT reaction of DMA3HF, without changing the photophysical properties, while introducing the electron-withdrawing group -CN can greatly improve the absorption of DMA3HF in the ultraviolet band, but hinders the occurrence of the ESIPT reaction.

1. Introduction
Excited-state intramolecular proton transfer (ESIPT), as one of the most basic modes of proton transfer in the photophysical and photochemical fields, is also a very common and important hydrogen bond dynamics behavior [1,2,3,4,5]. Since Weller first proposed the ESIPT mechanism in the middle of the last century [6], plenty of investigators have devoted themselves to experimental and theoretical studies on the ESIPT reaction, and derived various interesting research directions closely related to the ESIPT process [7,8,9,10,11,12,13,14,15,16,17,18]. The ESIPT reaction is essentially a photoisomerization behavior of molecules, which makes molecules exhibit double-fluorescence characteristics and a significant Stokes shift. Due to the large Stokes shift, there is almost no overlap between the absorption and emission of ESIPT-type molecules, which can avoid the interference of other fluorescent materials in the sample and the internal filtering effect. Moreover, on the basis of the uniquely high sensitivity of the ESIPT mechanism, it is easy to tune the fluorescence characteristics of ESIPT-type molecules. Based on these special properties, the molecules with ESIPT characteristics have been widely applied in fluorescent probes, ultraviolet filtering, chemosensors, etc. [19,20,21,22,23,24].
4′-N,N-dimethylamino-3-hydroxyflavone (DMA3HF)—an artificially synthesized fluorescent flavonoid derivative—has attracted wide attention in the fields of photophysics and chemistry due to its ESIPT characteristics, specific photophysical properties, and strong antioxidant activity [25,26,27]. Ghosh et al. [28] skillfully studied the ESIPT reaction of DMA3HF in sodium bis-ethylhexylsulfosuccinate (AOT), n-heptane, and water reverse micelle solutions by changing the ratio of water to AOT around DMA3HF encapsulated in nanovacuoles, and showed the influence of solvent polarity on the ESIPT process and photophysical properties of DMA3HF. Moreover, Das et al. [29] researched the ESIPT reaction efficiency of DMA3HF in the lyotropic liquid crystal (LLC) phase, thus exploring the hydrogen-bonding effects and polarity of LLC water molecules. Simultaneously, the effect of hydrogen bonds on the ESIPT reaction was clarified, and the slow ESIPT behavior observed experimentally in LLC was confirmed. Furukawa et al. [30] studied the effect of external electric fields on the ESIPT reaction and photophysical properties of DMA3HF in rigid polymethyl methacrylate (PMMA) films, and found that the excited-state dynamics of DMA3HF in a rigid environment are very different from those in solution. Furthermore, Ushakou et al. [31] explored the energetic characteristics of 3-hydroxyflavone (3-HF) and DMA3HF in enol and keto forms by comparing the spectroscopic properties of the two compounds in acetonitrile and ethyl acetate at different temperatures, which can provide reference for evaluating the reversibility of molecular proton transfer reactions. Nevertheless, the effect of substitution of different functional groups on the ESIPT mechanism and photophysical properties of the DMA3HF molecule is still lacking, and deserves further study.
For solving this issue, we designed two novel molecules (DMA3HF-CN and DMA3HF-NH2) by introducing the electron-withdrawing group cyano (-CN) and electron-donating group amino (-NH2) on the side of the proton acceptor, respectively (as can be seen in Figure 1). The -NH2 group as the classical strong electron-donating group with high activity, and the -CN group as the excellent hydrogen bond acceptor and strong electron-donating group, have been widely used in molecular modification. Since the stronger electron-donating group -N(CH3)2 is located at one end on the proton donor side, the introduction of functional groups at the other farthest end on the proton-acceptor side can more significantly cause the push/pull effect of -N(CH3)2 and its substituents on electrons, thereby tuning the ESIPT reaction of DMA3HF. The electron push/pull effect caused by the substitution of other carbons on the aromatic ring on the proton acceptor side is not as obvious as that of the carbon in the straight direction. Moreover, we predict that the introduction of the -CN group may weaken the electronegativity of the proton acceptor and inhibit the ESIPT process, while the introduction of the -NH2 group may promote the ESIPT reaction. In this work, the density functional theory (DFT) and time-dependent DFT (TD-DFT) methods were used to conduct comprehensive and detailed theoretical research on the ESIPT behavior as well as the photophysical properties of DMA3HF, DMA3HF-CN, and DMA3HF-NH2. The geometric parameters, corresponding infrared (IR) vibration spectra, natural bond orbital (NBO), fuzzy bond order (FBO), reduced density gradient (RDG) scatterplot, and topological parameters at the bond critical point (BCP) were obtained to explore the intensity changes of the intramolecular hydrogen bonds (IHBs), and also clarified the intensity relationships of the IHBs of DMA3HF, DMA3HF-CN, and DMA3HF-NH2. Moreover, the potential energy curves (PECs) were plotted to intuitively illustrate the degree of difficulty of proton transfer reaction, and the transition state (TS) structures along with the corresponding single-point energy (SPE) were obtained to calculate the exact values of the potential barriers. Furthermore, the corresponding electronic spectra of the three molecules were simulated to explore the influence of functional group substitution on the photophysical properties, and the frontier molecular orbitals (FMOs) associated with major transitions were also shown. We hope that this theoretical study on the ESIPT mechanism can provide some inspiration for the following purposeful search and synthesis of novel, high-quality, ESIPT-type fluorescent materials.
2. Computational Methods
In this study, the S0- and S1-state geometric configurations of the three molecules were fully optimized by the DFT and TD-DFT methods with the B3LYP functional and 6-311++G(d) basis set [32,33,34,35,36]. For keeping consistent with the experimental conditions, acetonitrile was selected as the solvent, and the polarizable continuum model (PCM) with the integral equation formalism variant (IEFPCM) was applied [37,38]. To accurately simulate the absorption and emission spectra, the fluorescence spectrum of DMA3HF in acetonitrile was calculated using seven different functionals, and the obtained results were compared with the experimental data [29], which indicated that the PBEPBE functional was the most suitable (see Table 1) [39,40,41,42,43,44]. Therefore, in this work, the absorption and fluorescence spectra for DMA3HF, DMA3HF-CN, and DMA3HF-NH2 were computed at the TD-DFT/PBEPBE/6-311++G(d) level. Moreover, the principal geometric parameters, IR vibration spectra, and NBO analysis were calculated based on the optimized geometric configurations [45,46,47]. Moreover, the FBO values [48], topological parameters at BCP [49], RDG scatterplots [50], IRI maps [51], and FMO maps of the three compounds were obtained using Multiwfn software (Version: 3.7, Beijing Kein Research Center for Natural Sciences, Beijing, China) and the VMD program (Version: 1.9.3, Theoretical and Computational Biophysics Group, Beckman Institute of the UIUC, Champaign-Urbana, IL, USA) [52,53]. The PECs of the three molecules in the S0 and S1 states were scanned by increasing the O1-H1 bond length with a fixed value. For the scans of the PECs, we performed restricted geometric optimizations for the three compounds. Furthermore, in order to obtain the exact values of the ESIPT reaction barriers, the corresponding TS structures of the three molecules were searched based on the quasi-Newton and synchronous transit (QST3) approach, and it was confirmed by vibration analysis that there was only one virtual mode corresponding to the proton transfer behavior [54]. We also observed the intrinsic reaction coordinate (IRC) curves to confirm that the TS structures we searched were correct [55]. All of the theoretical calculations in this work were achieved using the Gaussian 16 package (Version: Revision C.01, Gaussian, Inc., Wallingford, CT, USA) [56].
3. Results and Discussion
3.1. Optimized Geometric Structures and Infrared (IR) Vibrational Spectra Analysis
The geometric configurations of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 in enol and keto forms in different electronic states were optimized without any restrictions via the DFT and TD-DFT methods, and the main IHB geometric parameters are presented in Table 2. The comparison of geometric parameters related to IHBs (bond lengths and angles) can show the changes in IHBs’ intensity after the photoabsorption process. As listed, for enol configurations of DMA3HF and its derivatives, the O1-H1 bond length and ∠(O1-O1⋯O2) bond angle were all increased by the photoexcitation process, whereas the H1⋯O2 bond lengths were shortened. Concretely speaking, the O1-H1 bonds of the three molecules were separately elongated by 0.011 Å (DMA3HF) from 0.977 Å (S0) to 0.988 Å (S1), 0.009 Å (DMA3HF-CN) from 0.977 Å (S0) to 0.986 Å (S1), and 0.010 Å (DMA3HF-NH2) from 0.978 Å (S0) to 0.988 Å (S1). Similarly, the H1⋯O2 bonds were reduced by 0.116 Å, 0.111 Å, and 0.113 Å, respectively, from the S0 to S1 states. Meanwhile, the angles ∠(O1-H1⋯O2) increased from 117.886°, 117.187°, and 118.153° in the S0 state to 122.179°, 121.196°, and 122.390° in the S1 state, respectively. These results indicate that the IHBs of DMA3HF and its derivatives are all reinforced by the photoexcitation, which can promote the occurrence of the proton transfer process.
Monitoring the movement of stretching vibrational peaks related to IHBs in IR spectra is another available method to assess the changes in IHBs’ intensity [57,58]. Figure 2 depicts the calculated IR spectra of the three compounds at the S0 and S1 states, in the spectral range from 3200 cm−1 to 3700 cm−1, which corresponds to the region of O1-H1 stretching vibration peaks. It should be noted that the vibrational peaks of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 all display redshifts (S0→S1) to varying degrees, and the redshift magnitude of the three compounds is 188.89 cm−1, 165.14 cm−1, and 184.94 cm−1, respectively. Therefore, the O1-H1 bond strength is weakened and the attraction between H1 and O2 atoms is strengthened during the photoexcitation, which can promote the occurrence of the ESIPT reaction. The above conclusions are in great agreement with the results acquired from geometric structures.
3.2. Natural Bond Orbital (NBO) and Fuzzy Bond Order (FBO) Analysis
On the basis of the above discussion about geometric parameters and IR spectra, we can note that the IHBs are strengthened in the S1 state. Hydrogen bonds, as a weak interaction, are influenced by the charge over the correlation atoms, and the redistribution of atomic charge leads to changes in the intensity of IHBs. Therefore, the NBO population analysis was performed to quantitatively research the electronegativity of proton-donor and -acceptor moieties (O1 and O2 atoms). The charge distribution on O1 and O2 atoms of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 in the S0 and S1 states was calculated, and is summarized in Table 3. As shown, the negative charges located on the O1 atoms of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 decrease from the S0 to the S1 state, while those on the O2 atoms increase. That is, the attraction of O1 atoms to hydrogen protons is attenuated, while that of O2 atoms to protons is improved, corresponding to the elongation of O1-H1 bonds and the shortening of H1⋯O2 bonds under the photoexcitation.
Moreover, FBO analysis was introduced to further quantitatively represent the characteristics of O1-H1 bonds and H1⋯O2 bonds. Based on the division of fuzzy atomic space, FBO can directly reflect the degree of delocalization of electrons between two atomic spaces [59]. It is well established that the larger the magnitude of the FBO, the greater the bond strength. The FBO analysis results of O1-H1 bonds and H1⋯O2 bonds in the S0 and S1 states are exhibited in Table 4. We can see that, from the S0 to S1 states, the O1-H1 bond order values of the three compounds in enol form decreased, while the H1⋯O2 bond order values all increased. This implies that the ability of O1 atoms to bind the protons is reduced, while that of O2 atoms is enhanced, in the S1 state. Notably, the negative charge values distributed on the proton-acceptor O2 atoms of the three compounds in the S0 and S1 states can both be arranged in the following order: DMA3HF-CN < DMA3HF < DMA3HF-NH2. Meanwhile, the order of the H1⋯O2 FBO is the same. This means that, compared with DMA3HF, the substitution of the typical electron-withdrawing group (-CN) can attract away some electrons of the O2 atoms, thus weakening the ability of O2 atoms to capture the protons. Conversely, introducing the typical electron-donating group (-NH2) has the opposite effect.
3.3. Reduced Density Gradient (RDG) Scatterplot and Topology Analysis
Firstly, RDG analysis was selected to visually research the changes in the IHB strength of DMA3HF, DMA3HF-NH2, and DMA3HF-CN. The sign (λ2)ρ function obtained by multiplying the total electron density and the sign function of the second largest eigenvalue of the Hessian matrix for electron density was projected onto the RDG isosurface, and the intensity and type of weak interactions could be clearly seen [60]. When the RDG value of the scatter point is close to 0, the point corresponds to a weak interaction. For the sign (λ2)ρ function, the ρ-value can represent the strength of the interaction, and the sign (λ2) can denote the type of interaction. When the sign (λ2) is equal to −1, it represents attraction, while when the sign (λ2) is +1, it represents repulsion. By observing the molecular structure, we can know that the IHB is the strongest weak attractive interaction in the molecule. Therefore, we can confirm that the most negative spike corresponds to the IHB. In Figure 3a, the spikes representing the IHB of each molecule are circled, and the other negative spikes representing the other weak interactions are also marked (take DMA3HF-S0, for example). We compared the sign (λ2)ρ-values of IHBs’ spikes in the S0 and S1 states. It was found that the values of sign (λ2)ρ were decreased by 0.0067 a.u. (DMA3HF) from −0.0253 a.u. (S0) to −0.0320 a.u. (S1), 0.0062 a.u. (DMA3HF-CN) from −0.0239 a.u. (S0) to −0.0304 a.u. (S1), and 0.0066 a.u. (DMA3HF-NH2) from −0.0257 a.u. (S0) to −0.0323 a.u. (S1), implying that the IHBs are indeed enhanced in the S1 state. In addition, we also found that the IHB strength of DMA3HF-CN was weaker than that of the other two molecules. Incidentally, all of the chemical bonds and weak interaction regions of the three compounds in the S0 and the S1 states are shown by the interaction region indicator (IRI) plane color filling map and isosurface map, respectively, which can be seen in Figure 3b.
However, the sign (λ2)ρ-values of DMA3HF and DMA3HF-NH2 are very close, and cannot be distinguished only by the spikes in the RDG maps. Hence, we selected the atoms in molecules (AIM) theory to obtain the topological parameters at the bond critical points (BCPs) of the IHBs, and directly calculated the hydrogen bond energy (EHB) of the three compounds in the S0 and S1 states using empirical formulae [61,62]. The relevant parameters are listed in Table 5. Therein, the Laplacian of electron density ∇2ρ(r) values are positive, representing closed-shell interactions (corresponding to IHBs in this paper). In addition, the corresponding hydrogen bond was considered to be strong when the ρ(r) value at BCP was larger than 0.03 a.u., and the larger the value of ρ(r), the stronger the IHB. All of the relevant parameters indicate that the IHBs of the three molecules are strengthened in the S1 state. It is worth noting that the order of EHB for the three compounds is DMA3HF-CN < DMA3HF < DMA3HF-NH2, whether in the S0 or S1 state. This result directly illustrates that the substitution of the electron-donating group -NH2 on the proton-acceptor side can promote the ESIPT process; however, the substitution of the electron-withdrawing group -CN is able to inhibit the ESIPT behavior. The RDG and topology analysis provide proof for our previous conclusions acquired from FBO and NBO analyses.
3.4. Potential Energy Curves (PECs)
In order to further intuitively illustrate the degree of difficulty of proton transfer in the S0 and S1 states for DMA3HF, DMA3HF-CN, and DMA3HF-NH2, we scanned the PECs of the three molecules via lengthening the O1-H1 bonds from 1.0 Å to 2.0 Å, at a fixed step of 0.1 Å, and allowing all other degrees of freedom to relax freely towards the minimum energy [63,64], as shown in Figure 4. It can be seen that, for the three investigated molecules, the energy barriers for the forward proton transfer process in the S0 state were significantly larger than those in the S1 state, implying that the proton transfer behaviors are more likely to occur in the excited state. Moreover, the order of barriers for the three compounds in the S0 and S1 states are both DMA3HF-NH2 < DMA3HF < DMA3HF-CN, implying that the substitution of the -CN group at the proton-acceptor side impedes proton transfer, while the substitution of the -NH2 group promotes proton transfer, which is consistent with the results of NBO, FBO, RDG, and topology analyses.
Nevertheless, due to the limitation of scanning step length, the above PECs cannot accurately describe the reaction path of ESIPT. Therefore, it is necessary to search the transition state (TS) structures of molecules during proton transfer, and to accurately calculate the corresponding single-point energy (SPE). The SPE of all of the stable structures in the S1 state was calculated, as can be seen in Figure 5. As shown, the energy barriers for the ESIPT process were 7.1831 kcal/mol (for DMA3HF-NH2), 7.3212 kcal/mol (for DMA3HF), and 8.4198 kcal/mol (for DMA3HF-CN). This result confirms once again that the substitution of the -CN group on the proton-acceptor side hindered the ESIPT reaction, and the substitution of the -NH2 group would have the opposite effect. Furthermore, we drew the intrinsic reaction coordinate (IRC) curves based on the TS structures of the three molecules. As shown in Figure 6, the two ends of the IRC curves correspond to the enol and keto forms of the molecules, respectively, proving that the TS structures of the ESIPT reaction for which we searched were correct.
3.5. Electronic Spectra and Frontier Molecular Orbitals (FMOs)
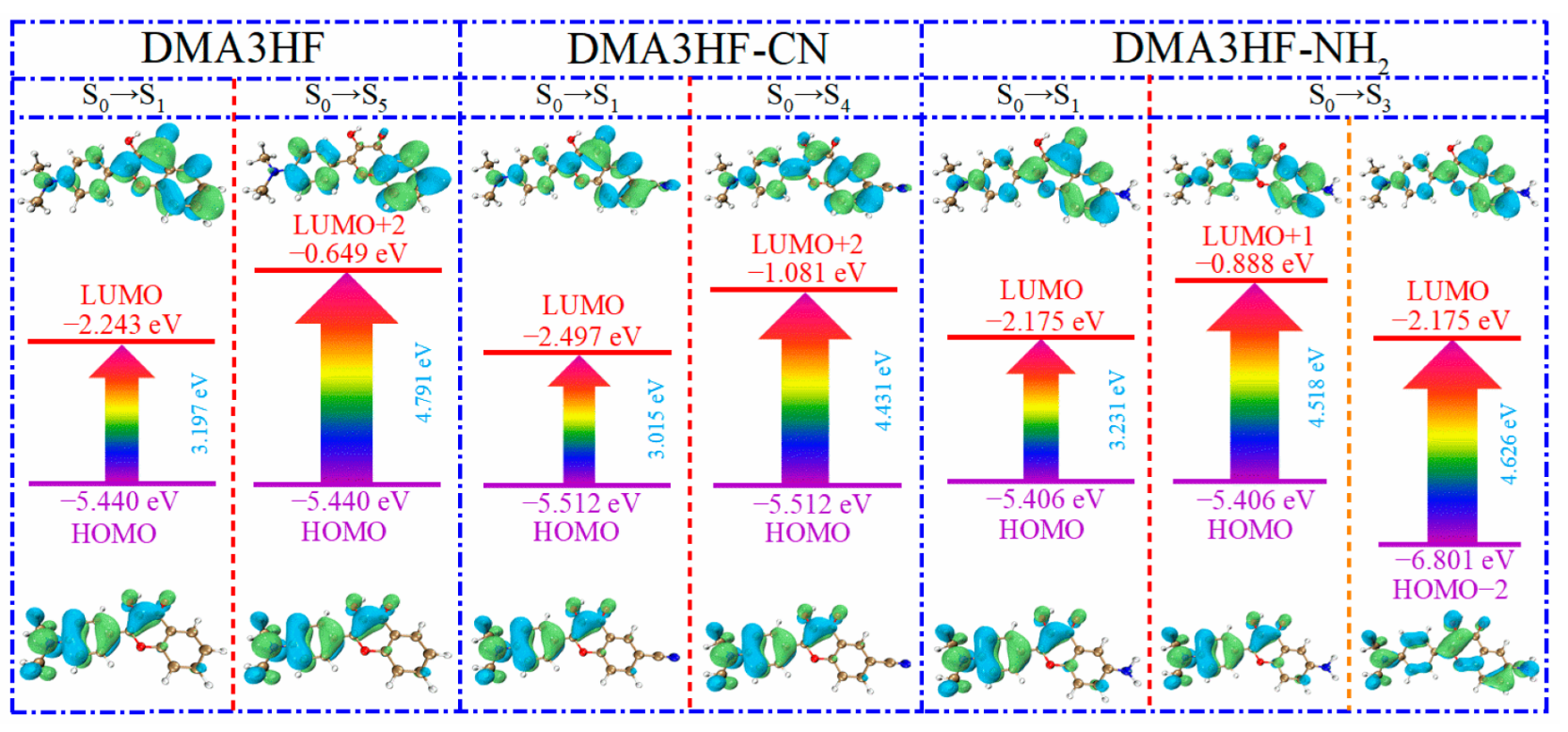
In this section, we explored the effects of two classical types of functional groups (-CN and -NH2) on the photophysical properties of DMA3HF. On the basis of the optimized ground- and excited-stated structures, the absorption and emission spectra of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 were simulated at the IEFPCM/TD-DFT/PBEPBE/6-311++G(d) level, and are displayed in Figure 7. Moreover, the transition properties (e.g., transition composition and oscillator strengths f) associated with the six low-lying absorption transitions (S1–S6) in acetonitrile are summarized in Table 6, and the fluorescence properties are listed in Table 7. As shown in Figure 7, the calculated fluorescence peaks of DMA3HF in the enol and keto forms are separately located at 532.94 nm and 586.30 nm, corresponding with the experimental values of 510 nm and 570 nm [29], and further indicating that the selected theoretical level is suitable for simulating the electronic spectra of DMA3HF, DMA3HF-CN, and DMA3HF-NH2. This also indirectly proves that the geometric structures optimized by the B3LYP functional are accurate. Notably, all three molecules possess obvious double-absorption peaks. Compared with the absorption spectra of DMA3HF, introducing the electron-donating group (-NH2) induced the absorption intensity of the dual peaks to increase to varying degrees, and the absorption peak located in the long-wavelength band exhibits a tiny blueshift of 2.63 nm. Moreover, the absorption peak of DMA3HF-CN in the long-wavelength band shows a redshift of 40.17 nm compared with that of DMA3HF, and the intensity of the absorption peak in the short-wavelength band increased obviously—even as high as the absorption peak in the long-wavelength band. As listed in Table 6, the absorption peaks of the three compounds in the long-wavelength band were ascribed to the S0→S1 transition, which was generated by the electronic transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). Furthermore, the absorption peaks of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 in the short-wavelength band correspond to the S0→S5 (HOMO→LUMO+2), S0→S4 (HOMO→LUMO+2), and S0→S3 transitions (HOMO-2→LUMO and HOMO→LUMO+1), respectively. Based on the transition properties of the three molecules, the corresponding FMO and energy gap diagrams are plotted in Figure 8. The occurrence of intramolecular charge transfer (ICT) behavior under the photoexcitation can be visually observed [65,66]. Notably, for the transition (S0→S1), the electronic cloud density distributed over the O1 atoms of the three compounds decreased, while that over the O2 atoms increased. That is, the ability of O1 atoms to attract protons was reduced, while that of O2 atoms is enhanced, which can advance the occurrence of the ESIPT reaction. Moreover, the peak shift phenomenon in the absorption spectra can be deduced from the energy gaps of the corresponding orbitals. It also can be seen that all of the absorption peaks of the three molecules originated from the π→π* transition of electrons.
Furthermore, we can see from Figure 7 and Table 7 that, compared with DMA3HF, the dual-fluorescence signals of DMA3HF-CN redshifted by 39.61 nm (enol form) and 16.71 nm (keto form). Similarly, the fluorescence peak of DMA3HF-NH2 in enol form blueshifted by 3.97 nm. It is noteworthy that the fluorescence intensities of the three molecules in the keto form were very strong. Moreover, the fluorescence peaks of the three molecules in keto form exhibited Stokes shifts of 79.67 nm (DMA3HF), 56.21 nm (DMA3HF-CN), and 81.59 nm (DMA3HF-NH2). The above results indicate that the substitution of the -CN group causes obvious redshift phenomena in the absorption and fluorescence spectra, and significantly enhances the absorption in the short-wavelength (ultraviolet) band. However, the substitution of the -NH2 group had no obvious effect on the photophysical properties of DMA3HF.
4. Conclusions
In this work, the influences of the electron-donating group -NH2 and electron-withdrawing group -CN on the ESIPT mechanism and photophysical properties of DMA3HF were comprehensively studied via DFT/TD-DFT methods. Based on the results obtained from the relevant geometric parameters, IR spectra, NBO charge population, FBO, RDG isosurface, and topology analysis, the excited-stated IHB strengthening mechanisms of DMA3HF and its two derivatives were confirmed. Moreover, according to the calculated PECs and TS structures corresponding to the ESIPT process, it was found that the substitution of -NH2 on the proton-acceptor side can promote the ESIPT process, while the substitution of -CN shows the opposite effect. In addition, from the simulated electronic spectra, it can be seen that DMA3HF and its two derivatives show strong fluorescence in the keto configuration compared with that in the enol configuration, and the introduction of -CN can greatly enhance the absorption intensity of DMA3HF in the ultraviolet band. In conclusion, the introduction of electron-donating and electron-withdrawing groups can regulate the ESIPT process of flavonoids and, thus, affect their optical properties. This theoretical investigation can provide valuable guidance in the experimental design and synthesis of efficient ESIPT-based fluorescence materials.
Author Contributions
Conceptualization, X.Y. and C.S. (Chaofan Sun); methodology, C.S. (Chaofan Sun); software, C.S. (Chaofan Sun); validation, X.Y. and C.S. (Changjiao Shang); formal analysis, X.Y. and Y.C.; investigation, C.S. (Changjiao Shang), Y.C., C.S. (Chaofan Sun), and J.C.; resources, J.C. and C.S. (Chaofan Sun); data curation, X.Y.; writing—original draft preparation, X.Y.; writing—review and editing, C.S. (Changjiao Shang), Y.C., J.C., and C.S. (Chaofan Sun); supervision, J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Training Program of Innovation and Entrepreneurship for Undergraduates (202110225159) and the Fundamental Research Funds for the Central Universities (2572020BC03).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wu, J.; Liu, W.; Ge, J.; Zhang, H.; Wang, P. New sensing mechanisms for design of fluorescent chemosensors emerging in recent years. Chem. Soc. Rev. 2011, 40, 3483–3495. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.E.; Park, S.Y. Advanced Organic Optoelectronic Materials: Harnessing Excited-State Intramolecular Proton Transfer (ESIPT) Process. Adv. Mater. 2011, 23, 3615–3642. [Google Scholar] [CrossRef]
- Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Excited state intramolecular proton transfer (ESIPT): From principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. [Google Scholar] [CrossRef] [PubMed]
- Padalkar, V.S.; Seki, S. Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters. Chem. Soc. Rev. 2016, 45, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, A.C.; Wu, L.L.; Han, H.H.; Bull, S.D.; He, X.P.; James, T.D.; Sessler, J.L.; Tang, B.Z.; Tian, H.; Yoon, J. Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents. Chem. Soc. Rev. 2018, 47, 8842–8880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, A. Über die Fluoreszenz der Salizylsäure und verwandter Verbindungen. Naturwissenschaften 1955, 42, 175–176. [Google Scholar] [CrossRef]
- Chen, L.; Ye, J.W.; Wang, H.P.; Pan, M.; Yin, S.Y.; Wei, Z.W.; Zhang, L.Y.; Wu, K.; Fan, Y.N.; Su, C.Y. Ultrafast water sensing and thermal imaging by a metal-organic framework with switchable luminescence. Nat. Commun. 2017, 8, 15985. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, B.F.; Zhou, Q.; Zhang, X.; Li, B.; Su, X.; Shi, Y. Enhancing fluorescence of benzimidazole derivative via solvent-regulated ESIPT and TICT process: A TDDFT study. Spectrochim. Acta Part A 2021, 258, 119862. [Google Scholar] [CrossRef]
- Cao, B.F.; Han, J.H.; Zhou, Q.; Sun, C.F.; Li, Y.; Li, B.; Yin, H.; Shi, Y. Skillfully tuning 1-hydroxy-9H-fluoren-9-one forward-backward ESIPT processes by introducing electron-withdrawing groups: A theoretical exploration. J. Mol. Liq. 2020, 303, 112627. [Google Scholar] [CrossRef]
- Luo, X.; Shi, W.; Yang, Y.F.; Song, Y.Z.; Li, Y.Q. Systematic theoretical investigation of two novel molecules BtyC-1 and BtyC-2 based on ESIPT mechanism. Spectrochim. Acta Part A 2021, 258, 119810. [Google Scholar] [CrossRef]
- Zhao, G.J.; Shi, W.; Yang, Y.F.; Ding, Y.; Li, Y.Q. Substituent Effects on Excited-State Intramolecular Proton Transfer Reaction of 2-Aryloxazoline Derivatives. J. Phys. Chem. A 2021, 125, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.P.; Jia, M.; Zhang, Q.L.; Wang, Y.S. Modulating O-H-based excited-state intramolecular proton transfer by alkyl-substitutions at various positions of 1-hydroxy-11H-benzo b fluoren-11-one. J. Lumin. 2020, 219, 116913. [Google Scholar] [CrossRef]
- Yang, D.P.; Zhang, T.J.; Song, X.Y.; Gao, H.Y. Is excited state intramolecular proton transfer frustrated in 10-hydroxy-11H-benzo b fluoren-11-one? Spectrochim. Acta Part A Spectrosc. 2020, 228, 117734. [Google Scholar] [CrossRef] [PubMed]
- Daengngern, R.; Kungwan, N. Electronic and photophysical properties of 2-(2′-hydroxyphenyl)benzoxazole and its derivatives enhancing in the excited-state intramolecular proton transfer processes: A TD-DFT study on substitution effect. J. Lumin. 2015, 167, 132–139. [Google Scholar] [CrossRef]
- Daengngern, R.; Prommin, C.; Rungrotmongkol, T.; Promarak, V.; Wolschann, P.; Kungwan, N. Theoretical investigation of 2-(iminomethyl) phenol in the gas phase as a prototype of ultrafast excited-state intramolecular proton transfer. Chem. Phys. Lett. 2016, 657, 113–118. [Google Scholar] [CrossRef]
- Kanlayakan, N.; Kerdpol, K.; Prommin, C.; Salaeh, R.; Chansen, W.; Sattayanon, C.; Kungwan, N. Effects of different proton donor and acceptor groups on excited-state intramolecular proton transfers of amino-type and hydroxy-type hydrogen-bonding molecules: Theoretical insights. New J. Chem. 2017, 41, 8761–8771. [Google Scholar] [CrossRef]
- Jia, L.F.; Wang, F.; Liu, Y.F. Solvent effects on excited state intramolecular proton transfer mechanism in 4-(N,N-dimethylamino)-3-hydroxyflavone. Org. Electron. 2018, 57, 292–297. [Google Scholar] [CrossRef]
- Li, C.Z.; Li, D.L.; Ma, C.; Liu, Y.F. DFT-TDDFT investigation of excited-state intramolecular proton transfer in 2-(2′-hydroxyphenyl)benzimidazole derivatives: Effects of electron acceptor and donor groups. J. Mol. Liq. 2016, 224, 83–88. [Google Scholar] [CrossRef]
- Yang, L.; Yang, N.; Gu, P.; Wang, C.; Li, B.; Zhang, Y.; Ji, L.; He, G. A novel flavone-based ESIPT ratiometric fluorescent probe for selective sensing and imaging of hydrogen polysulfides. Spectrochim. Acta Part A 2022, 271, 120962. [Google Scholar] [CrossRef] [PubMed]
- Holt, E.L.; Krokidi, K.M.; Turner, M.A.P.; Mishra, P.; Zwier, T.S.; Rodrigues, N.D.N.; Stavros, V.G. Insights into the photoprotection mechanism of the UV filter homosalate. Phys. Chem. Chem. Phys. 2020, 22, 15509–15519. [Google Scholar] [CrossRef]
- Feng, W.X.; Fu, G.R.; Huang, Y.J.; Zhao, Y.; Yan, H.X.; Lu, X.Q. ESIPT-capable Eu3+-metallopolymer with colour-tunable emission for selective visual sensing of Zn2+ ion. J. Mater. Chem. C 2022, 10, 1090–1096. [Google Scholar] [CrossRef]
- Cao, Y.J.; Yu, X.R.; Sun, C.F.; Cui, J.A. Theoretical Investigation on the ESIPT Process and Detection Mechanism for Dual-Proton Type Fluorescent Probe. Int. J. Mol. Sci. 2022, 23, 2132. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, D. A Selective Fluorescence Turn-On Probe for the Detection of DCNP (Nerve Agent Tabun Simulant). Materials 2019, 12, 2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.L.; Liu, Y.F. A theoretical investigation on ESIPT process of a red-emitting ratiometric fluorescent probe and its fluorescent detection mechanism for cyanide anion. J. Ind. Eng. Chem. 2021, 99, 126–133. [Google Scholar] [CrossRef]
- Kenfack, C.A.; Klymchenko, A.S.; Duportail, G.; Burger, A.; Mely, Y. Ab initio study of the solvent H-bonding effect on ESIPT reaction and electronic transitions of 3-hydroxychromone derivatives. Phys. Chem. Chem. Phys. 2012, 14, 8910–8918. [Google Scholar] [CrossRef]
- Zhu, A.; Wang, B.; White, J.O.; Drickamer, H.G. The effects of pressure on the intramolecular proton transfer and charge transfer of 4′-N-dimethylamino-3-hydroxyflavone. J. Phys. Chem. B 2004, 108, 891–894. [Google Scholar] [CrossRef]
- Karmakar, A.; Mallick, T.; Fouzder, C.; Mukhuty, A.; Mondal, S.; Pramanik, A.; Kundu, R.; Mandal, D.; Begum, N.A. Unfolding the Role of a Flavone-Based Fluorescent Antioxidant towards the Misfolding of Amyloid Proteins: An Endeavour to Probe Amyloid Aggregation. J. Phys. Chem. B 2020, 124, 11133–11144. [Google Scholar] [CrossRef]
- Ghosh, D.; Batuta, S.; Begum, N.A.; Mandal, D. Unusually slow intramolecular proton transfer dynamics of 4′-N,N-dimethylamino-3-hydroxyflavone in high n-alcohols: Involvement of solvent relaxation. Photochem. Photobiol. Sci. 2016, 15, 266–277. [Google Scholar] [CrossRef]
- Das, K.; Sappati, S.; Bisht, G.S.; Hazra, P. Proton-Coupled Electron Transfer in the Aqueous Nanochannels of Lyotropic Liquid Crystals: Interplay of H-Bonding and Polarity Effects. J. Phys. Chem. Lett. 2021, 12, 2651–2659. [Google Scholar] [CrossRef]
- Furukawa, K.; Hino, K.; Yamamoto, N.; Awasthi, K.; Nakabayashi, T.; Ohta, N.; Sekiya, H. External Electric Field Effects on Excited-State Intramolecular Proton Transfer in 4′-N,N-Dimethylamino-3-hydroxyflavone in Poly(methyl methacrylate) Films. J. Phys. Chem. A 2015, 119, 9599–9608. [Google Scholar] [CrossRef]
- Ushakou, D.V.; Tomin, V.I. Spectroscopic methods for the study of energetic characteristics of the normal and photoproduct forms of 3-hydroxyflavones. Spectrochim. Acta Part A 2018, 204, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.W.; Han, K. Unraveling the Detailed Mechanism of Excited-State Proton Transfer. Acc. Chem. Res. 2018, 51, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Jacquemin, D.; Wathelet, V.; Perpete, E.A.; Adamo, C. Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Klamt, A.; Moya, C.; Palomar, J. A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach. J. Chem. Theory Comput. 2015, 11, 4220–4225. [Google Scholar] [CrossRef] [Green Version]
- Roch, L.M.; Baldridge, K.K. General optimization procedure towards the design of a new family of minimal parameter spin-component-scaled double-hybrid density functional theory. Phys. Chem. Chem. Phys. 2017, 19, 26191–26200. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Son, H.J.; Choe, J.I. mPW1PW91 study for conformational isomers of methylene bridge-monosubstituted tetramethoxycalix 4 arenes. J. Ind. Eng. Chem. 2014, 20, 3276–3282. [Google Scholar] [CrossRef]
- Ramalingam, S.; Periandy, S.; Mohan, S. Vibrational spectroscopy (FTIR and FTRaman) investigation using ab initio (HF) and DFT (B3LYP and B3PW91) analysis on the structure of 2-amino pyridine. Spectrochim. Acta Part A 2010, 77, 73–81. [Google Scholar] [CrossRef]
- Babu, P.D.S.; Periandy, S.; Mohan, S.; Ramalingam, S.; Jayaprakash, B.G. Molecular structure and vibrational investigation of benzenesulfonic acid methyl ester using DFT (LSDA, B3LYP, B3PW91 and MPW1PW91) theory calculations. Spectrochim. Acta Part A 2011, 78, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Khajehzadeh, M.; Moghadam, M. Molecular structure, FT IR, NMR, UV, NBO and HOMO-LUMO of 1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran- 5-carbonitrile by DFT/B3LYP and PBEPBE methods with LanL2DZ and 6-311++G(d,2p) basis sets. Spectrochim. Acta Part A 2017, 180, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.J.; Wang, L.L.; Cao, Y.J.; Yu, X.R.; Li, Y.Z.; Sun, C.F.; Cui, J.G. Is it possible to switch ESIPT-channel of hydroxyanthraquinones with the strategy of modifying electronic groups? J. Mol. Liq. 2022, 347, 118343. [Google Scholar] [CrossRef]
- Sun, C.F.; Li, H.; Yin, H.; Li, Y.Z.; Shi, Y. Effects of the cyano substitution at different positions on the ESIPT properties of alizarin: A DFT/TD-DFT investigation. J. Mol. Liq. 2018, 269, 650–656. [Google Scholar] [CrossRef]
- Sun, C.F.; Zhao, H.F.; Liu, X.C.; Yin, H.; Shi, Y. Tunable ESIPT reaction and antioxidant activities of 3-hydroxyflavone and its derivatives by altering atomic electronegativity. Org. Chem. Front. 2018, 5, 3435–3442. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Bond Order Analysis Based on the Laplacian of Electron Density in Fuzzy Overlap Space. J. Phys. Chem. A 2013, 117, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Duran, M.; Sola, M.; Silvi, B. Theoretical evaluation of electron delocalization in aromatic molecules by means of atoms in molecules (AIM) and electron localization function (ELF) topological approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W.T. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chemistry-Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bao, J.L.; Truhlar, D.G. Variational transition state theory: Theoretical framework and recent developments. Chem. Soc. Rev. 2017, 46, 7548–7596. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Harabuchi, Y.; Ono, Y.; Taketsugu, T.; Morokuma, K. Intrinsic Reaction Coordinate: Calculation, Bifurcation, and Automated Search. Int. J. Quantum Chem. 2015, 115, 258–269. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Su, X.; Zhou, Q.; Li, Y.; Cao, B.F.; Li, B.; Zhang, X.; Yin, H.; Shi, Y. Revised the excited-state intramolecular proton transfer direction of the BTHMB molecule: A theoretical study. Spectrochim. Acta Part A 2021, 249, 119327. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.J.; Cao, Y.J.; Sun, C.F.; Li, Y.Z. Unveiling the influence of atomic electronegativity on the double ESIPT processes of uralenol: A theoretical study. Spectrochim. Acta Part A 2022, 268, 120660. [Google Scholar] [CrossRef]
- Matito, E.; Poater, J.; Sola, M.; Duran, M.; Salvador, P. Comparison of the AIM delocalization index and the Mayer and Fuzzy atom bond orders. J. Phys. Chem. A 2005, 109, 9904–9910. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.C.; Contreras-Garcia, J.; Henon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Nakanishi, W.; Hayashi, S.; Narahara, K. Atoms-in-Molecules Dual Parameter Analysis of Weak to Strong Interactions: Behaviors of Electronic Energy Densities versus Laplacian of Electron Densities at Bond Critical Points. J. Phys. Chem. A 2008, 112, 13593–13599. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.F.; Su, X.; Zhou, Q.; Shi, Y. Regular tuning of the ESIPT reaction of 3-hydroxychromone-based derivatives by substitution of functional groups. Org. Chem. Front. 2019, 6, 3093–3100. [Google Scholar] [CrossRef]
- Ma, Y.Z.; Yang, Y.F.; Lan, R.F.; Li, Y.Q. Effect of Different Substituted Groups on Excited-State Intramolecular Proton Transfer of 1-(Acylamino)-anthraquinons. J. Phys. Chem. C 2017, 121, 14779–14786. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural changes accompanying intramolecular electron transfer: Focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 2003, 103, 3899–4031. [Google Scholar] [CrossRef]
- Peng, X.J.; Song, F.L.; Lu, E.; Wang, Y.N.; Zhou, W.; Fan, J.L.; Gao, Y.L. Heptamethine cyanine dyes with a large stokes shift and strong fluorescence: A paradigm for excited-state intramolecular charge transfer. J. Am. Chem. Soc. 2005, 127, 4170–4171. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Molecular structures of DMA3HF and its derivatives in (a) enol and (b) keto forms.

Figure 2.
Simulated IR spectra of the three compounds in acetonitrile at the spectral region of the O1–H1 stretching band.
Figure 2.
Simulated IR spectra of the three compounds in acetonitrile at the spectral region of the O1–H1 stretching band.
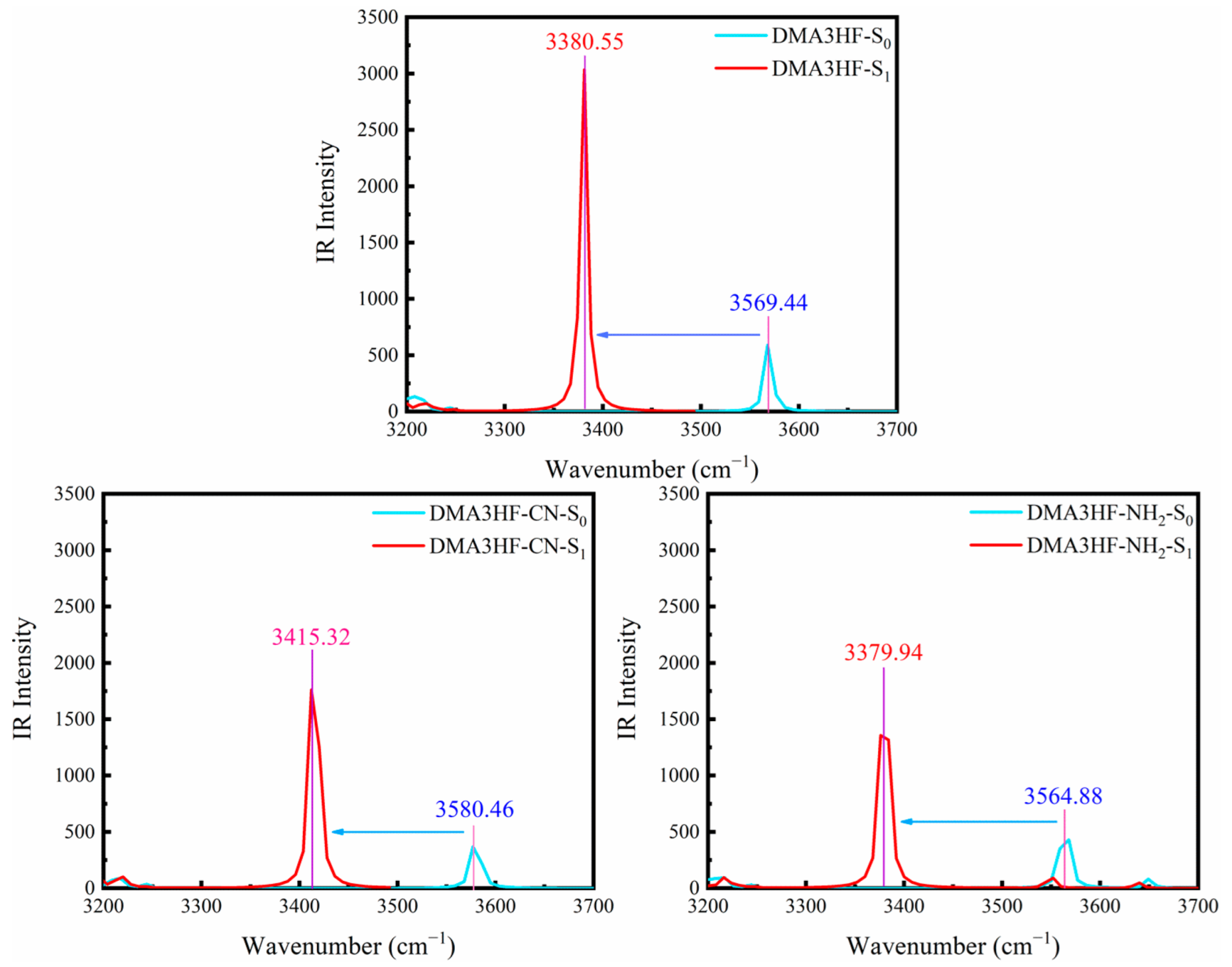
Figure 3.
RDG scatterplots and IRI maps of DMA3HF and its two derivatives in the S0 and S1 states: (a) the RDG versus sign (λ2)ρ scatterplots of the three compounds; (b) the IRI plane color filling map in the S0 state and IRI isosurface map in the S1 state of the three compounds.
Figure 3.
RDG scatterplots and IRI maps of DMA3HF and its two derivatives in the S0 and S1 states: (a) the RDG versus sign (λ2)ρ scatterplots of the three compounds; (b) the IRI plane color filling map in the S0 state and IRI isosurface map in the S1 state of the three compounds.
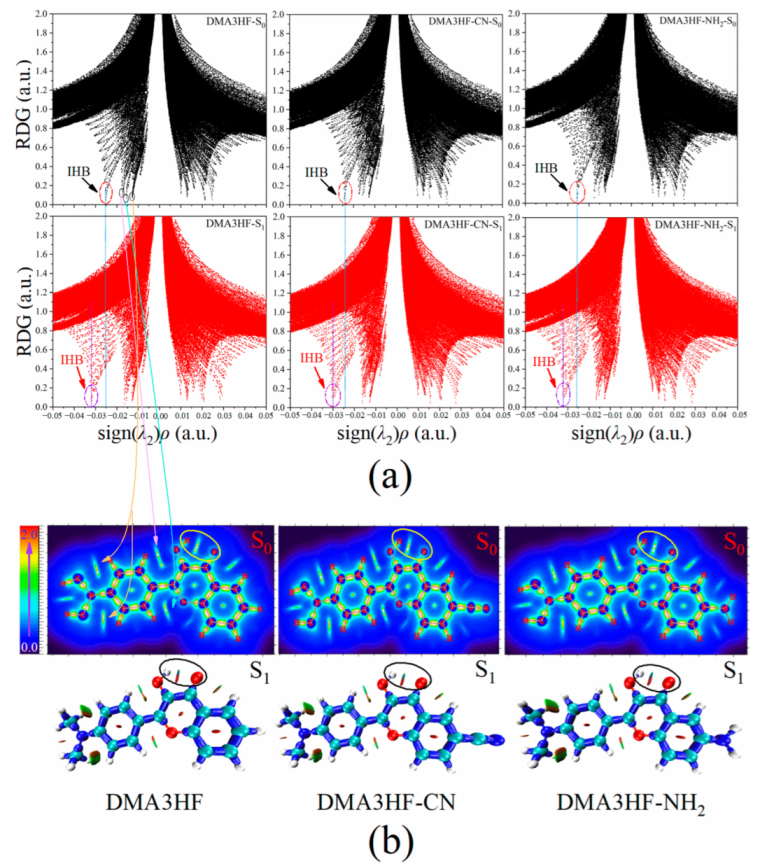
Figure 4.
Scanned PECs of the three molecules in the S0 and S1 states.

Figure 5.
ESIPT reaction energy profiles for DMA3HF and its two derivatives.
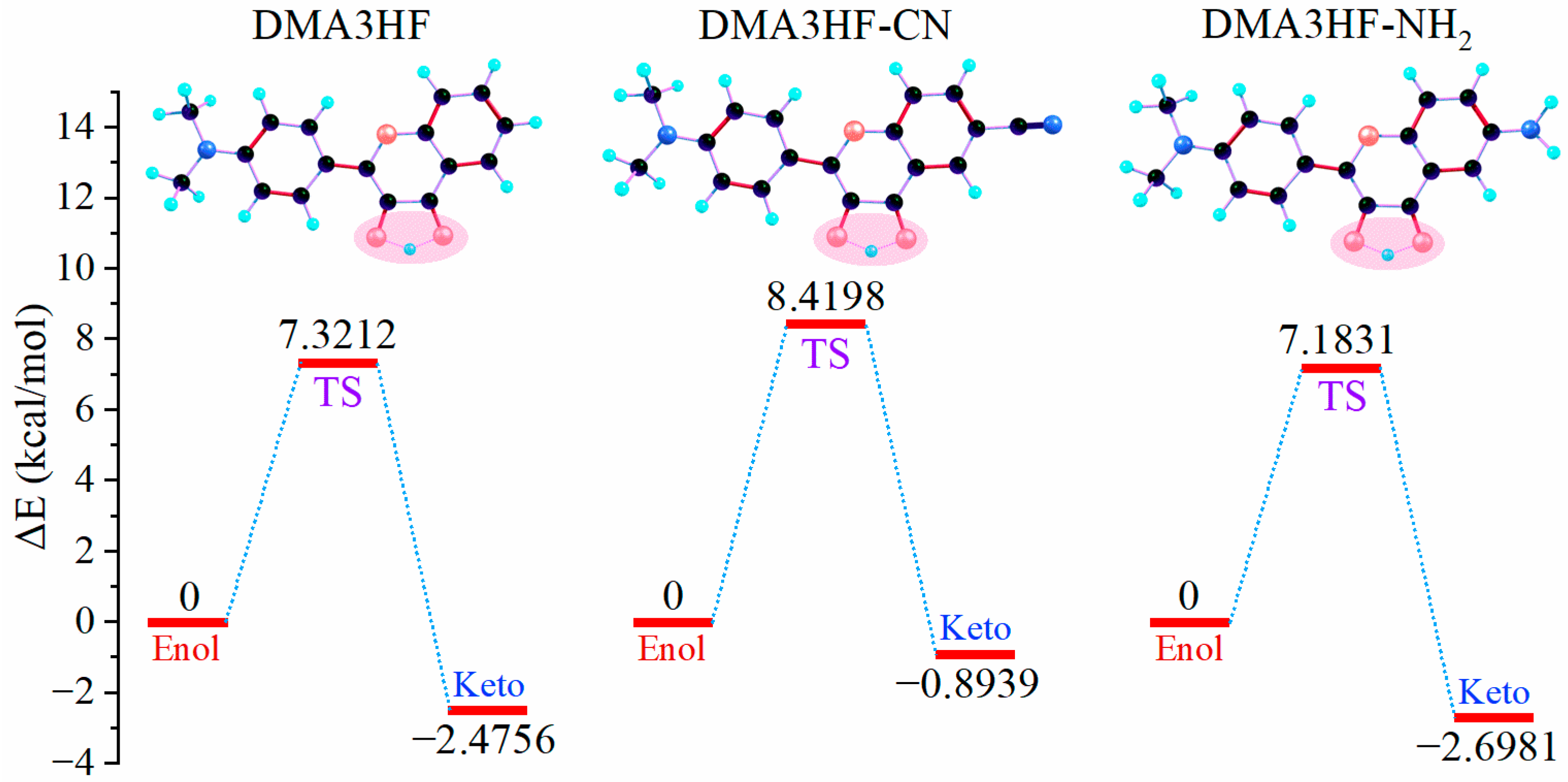
Figure 6.
IRC curves scanned based on the TS structures of the three molecules.
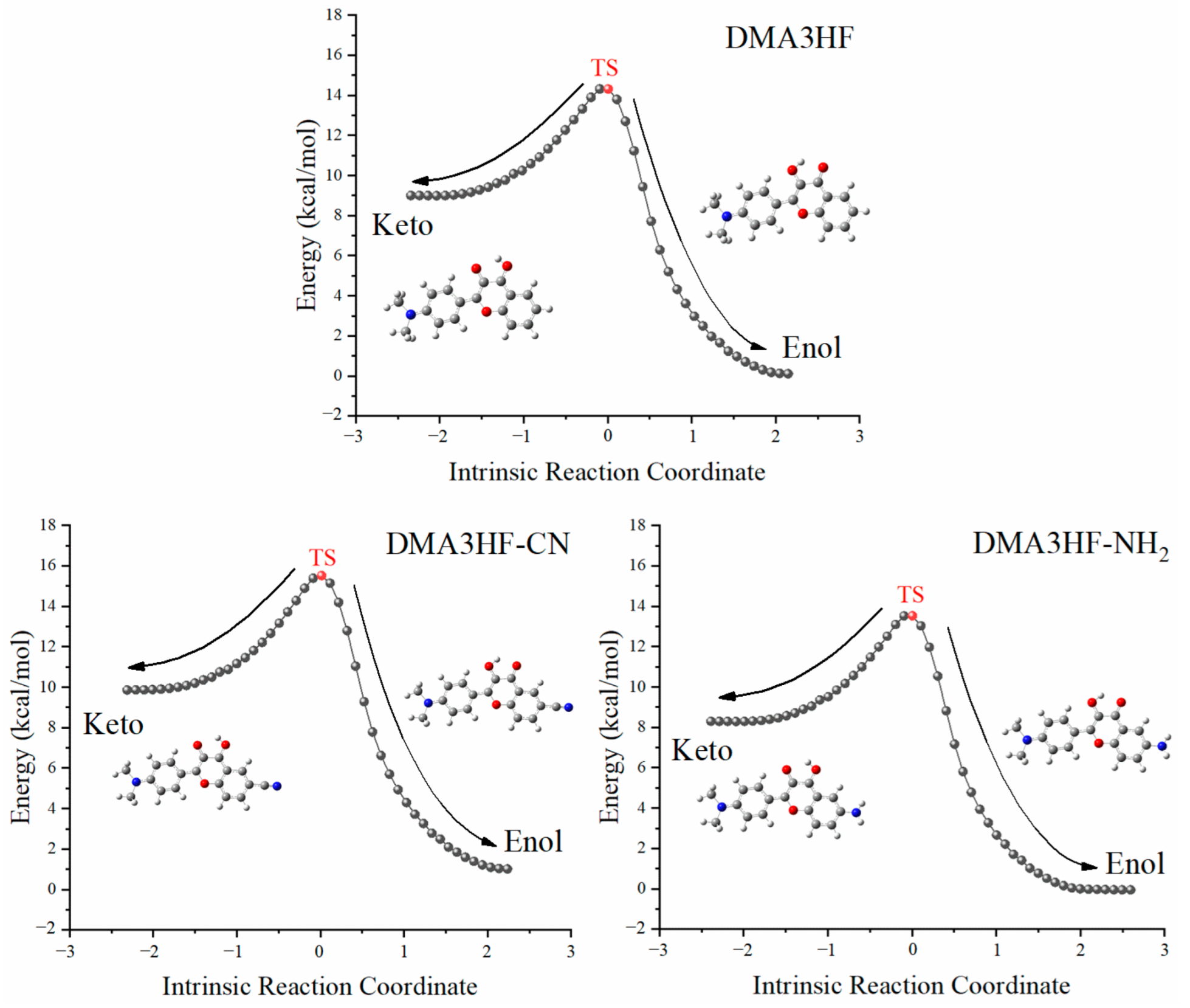
Figure 7.
Simulated absorption and fluorescence spectra of the three molecules in the enol and keto forms in acetonitrile.
Figure 7.
Simulated absorption and fluorescence spectra of the three molecules in the enol and keto forms in acetonitrile.

Figure 8.
Frontier molecular orbitals and energy gaps of the three compounds.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated fluorescence peaks (nm) of DMA3HF in acetonitrile, obtained via the TD-DFT method with seven different functionals.
Table 1.
Calculated fluorescence peaks (nm) of DMA3HF in acetonitrile, obtained via the TD-DFT method with seven different functionals.
| PBEPBE | B3PW91 | Cam-B3LYP | B3LYP | M062x | mPW1PW91 | ωB97XD | Exp a | |
|---|---|---|---|---|---|---|---|---|
| λflu 1 | 532.94 | 456.48 | 396.43 | 458.03 | 396.64 | 441.74 | 388.21 | 510 |
| λflu 2 | 586.30 | 545.90 | 520.40 | 547.40 | 513.09 | 537.39 | 518.42 | 570 |
a Maximum fluorescence peaks in the experiment.
Table 2.
Calculated bond lengths (Å) and bond angles (°) related to the IHBs of DMA3HF and its derivatives in enol and keto forms in the S0 and S1 states, respectively.
Table 2.
Calculated bond lengths (Å) and bond angles (°) related to the IHBs of DMA3HF and its derivatives in enol and keto forms in the S0 and S1 states, respectively.
| State | O1-H1 | H1-O2 | ∠(O1-H1⋯O2) | |
|---|---|---|---|---|
| DMA3HF-enol | S0 | 0.977 | 2.025 | 117.886 |
| S1 | 0.988 | 1.909 | 122.179 | |
| DMA3HF-keto | S0 | 1.936 | 0.988 | 120.425 |
| S1 | 2.009 | 0.981 | 117.761 | |
| DMA3HF-CN-enol | S0 | 0.977 | 2.044 | 117.187 |
| S1 | 0.986 | 1.933 | 121.196 | |
| DMA3HF-CN-keto | S0 | 1.960 | 0.987 | 119.440 |
| S1 | 2.018 | 0.981 | 117.213 | |
| DMA3HF-NH2-enol | S0 | 0.978 | 2.019 | 118.153 |
| S1 | 0.988 | 1.906 | 122.390 | |
| DMA3HF-NH2-keto | S0 | 1.934 | 0.988 | 120.556 |
| S1 | 2.004 | 0.981 | 118.011 |
Table 3.
Calculated distribution of NBO charges (a.u.) on the O1 and O2 atoms of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 in the S0 and S1 states.
Table 3.
Calculated distribution of NBO charges (a.u.) on the O1 and O2 atoms of DMA3HF, DMA3HF-CN, and DMA3HF-NH2 in the S0 and S1 states.
| DMA3HF | DMA3HF-CN | DMA3HF-NH2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| State/Δ | S0 | S1 | Δ | S0 | S1 | Δ | S0 | S1 | Δ |
| O1 | −0.6917 | −0.6580 | −0.0337 | −0.6860 | −0.6540 | −0.0320 | −0.6947 | −0.6639 | −0.0308 |
| O2 | −0.6948 | −0.7646 | +0.0698 | −0.6819 | −0.7413 | +0.0594 | −0.7024 | −0.7741 | +0.0717 |
Δ: Difference in NBO charges between the S0 and S1 states; positive values represent increases and negative values represent decreases (S0→S1).
Table 4.
Obtained fuzzy bond order related to the ESIPT process.
| State | FBO (O1-H1) | FBO (H1⋯O2) | |
|---|---|---|---|
| DMA3HF-enol | S0 | 0.75626 | 0.06129 |
| S1 | 0.72953 | 0.07995 | |
| DMA3HF-keto | S0 | 0.08263 | 0.72416 |
| S1 | 0.07044 | 0.74092 | |
| DMA3HF-CN-enol | S0 | 0.75760 | 0.05773 |
| S1 | 0.73284 | 0.07482 | |
| DMA3HF-CN-keto | S0 | 0.07747 | 0.72767 |
| S1 | 0.06834 | 0.73979 | |
| DMA3HF-NH2-enol | S0 | 0.75591 | 0.06242 |
| S1 | 0.72957 | 0.08095 | |
| DMA3HF-NH2-keto | S0 | 0.08309 | 0.72530 |
| S1 | 0.07136 | 0.74132 |
Table 5.
Calculated topological parameters at BCPs related to the IHBs of the three molecules in enol and keto forms in the S0 and S1 states.
Table 5.
Calculated topological parameters at BCPs related to the IHBs of the three molecules in enol and keto forms in the S0 and S1 states.
| ρ(r) α | ∇2ρ(r) β | V(r) γ | G(r) δ | H(r) ε | ELF ζ | EHB η | |
|---|---|---|---|---|---|---|---|
| DMA3HF-enol-S0 | 0.0253 | 0.1026 | −0.0204 | 0.0230 | 0.0027 | 0.0689 | −4.9016 |
| DMA3HF-enol-S1 | 0.0320 | 0.1209 | −0.0270 | 0.0286 | 0.0016 | 0.0950 | −6.3963 |
| DMA3HF-keto-S0 | 0.0310 | 0.1133 | −0.0254 | 0.0268 | 0.0015 | 0.0964 | −6.1732 |
| DMA3HF-keto-S1 | 0.0267 | 0.1009 | −0.0212 | 0.0232 | 0.0020 | 0.0805 | −5.2139 |
| DMA3HF-CN-enol-S0 | 0.0239 | 0.1241 | −0.0222 | 0.0266 | 0.0044 | 0.0439 | −4.5893 |
| DMA3HF-CN-enol-S1 | 0.0304 | 0.1168 | −0.0253 | 0.0272 | 0.0020 | 0.0887 | −6.0393 |
| DMA3HF-CN-keto-S0 | 0.0294 | 0.1095 | −0.0238 | 0.0256 | 0.0018 | 0.0899 | −5.8163 |
| DMA3HF-CN-keto-S1 | 0.0262 | 0.1000 | −0.0207 | 0.0229 | 0.0022 | 0.0781 | −5.1024 |
| DMA3HF-NH2-enol-S0 | 0.0257 | 0.1035 | −0.0207 | 0.0233 | 0.0026 | 0.0704 | −4.9909 |
| DMA3HF-NH2-enol-S1 | 0.0323 | 0.1215 | −0.0272 | 0.0288 | 0.0016 | 0.0959 | −6.4632 |
| DMA3HF-NH2-keto-S0 | 0.0311 | 0.1137 | −0.0255 | 0.0270 | 0.0015 | 0.0969 | −6.1955 |
| DMA3HF-NH2-keto-S1 | 0.0270 | 0.1017 | −0.0214 | 0.0234 | 0.0020 | 0.0817 | −5.2809 |
α: Density of all electrons (a.u.); β: Laplacian of electron density (a.u.); γ: potential energy density (a.u.); δ: Lagrangian kinetic energy (a.u.); ε: energy density (a.u.); ζ: electron localization function (a.u.); η: hydrogen bond energy (kcal/mol), EHB = –223.08ρ(r) + 0.7423.
Table 6.
Calculated transition properties of the three compounds in acetonitrile.
| State | λabs (nm) | Contribution MO a | Strength f | |
|---|---|---|---|---|
| DMA3HF | S1 | 506.63 | (68.325%) H→L | 0.5655 |
| S2 | 369.48 | (56.740%) H→L + 1 (34.285%) H→L + 2 | 0.1037 | |
| S3 | 366.37 | (67.241%) H-1→L | 0.0431 | |
| S4 | 344.55 | (70.680%) H-2→L | 0.0000 | |
| S5 | 344.03 | (55.193%) H→L + 2 | 0.2958 | |
| S6 | 329.47 | (45.859%) H-3→L (46.421%) H→L + 3 | 0.0086 | |
| DMA3HF-CN | S1 | 546.80 | (67.535%) H→L | 0.5329 |
| S2 | 504.39 | (69.415%) H→L + 1 | 0.0359 | |
| S3 | 383.18 | (65.797%) H-1→L | 0.0241 | |
| S4 | 359.31 | (63.652%) H→L + 2 | 0.5336 | |
| S5 | 355.10 | (70.573%) H-2→L | 0.0000 | |
| S6 | 343.13 | (66.814%) H-3→L | 0.0040 | |
| DMA3HF-NH2 | S1 | 504.00 | (68.324%) H→L | 0.5836 |
| S2 | 445.18 | (67.881%) H-1→L | 0.0474 | |
| S3 | 353.16 | (47.579%) H-2→L (46.050%) H→L + 1 | 0.3820 | |
| S4 | 343.13 | (69.751%) H-3→L | 0.0051 | |
| S5 | 339.41 | (45.346%) H-2→L (45.640%) H→L + 1 | 0.1166 | |
| S6 | 336.45 | (58.989%) H→L + 2 (31.026%) H→L + 3 | 0.0147 |
a: Molecular Orbitals; H: the highest occupied molecular orbital (HOMO); L: the lowest unoccupied molecular orbital (LUMO).
Table 7.
Calculated fluorescence properties of the three molecules in the enol and keto forms in acetonitrile.
Table 7.
Calculated fluorescence properties of the three molecules in the enol and keto forms in acetonitrile.
| State | Eflu (eV) | λflu (nm) | Contribution MO a | Strength f | |
|---|---|---|---|---|---|
| DMA3HF-enol | S1 | 2.3264 | 532.94 | H→L (68.670%) | 0.6049 |
| DMA3HF-keto | S1 | 2.1147 | 586.30 | H→L (71.559%) | 0.7774 |
| DMA3HF-CN-enol | S1 | 2.1655 | 572.55 | H→L (68.886%) | 0.5805 |
| DMA3HF-CN-keto | S1 | 2.0561 | 603.01 | H→L (70.403%) | 0.7765 |
| DMA3HF-NH2-enol | S1 | 2.3439 | 528.97 | H→L (68.663%) | 0.6291 |
| DMA3HF-NH2-keto | S1 | 2.1173 | 585.59 | H→L (71.017%) | 0.7374 |
a: Molecular Orbitals; H: the highest occupied molecular orbital (HOMO); L: the lowest unoccupied molecular orbital (LUMO).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yu, X.; Shang, C.; Cao, Y.; Cui, J.; Sun, C. A DFT/TD-DFT Study on the ESIPT-Type Flavonoid Derivatives with High Emission Intensity. Materials 2022, 15, 2896. https://doi.org/10.3390/ma15082896
AMA Style
Yu X, Shang C, Cao Y, Cui J, Sun C. A DFT/TD-DFT Study on the ESIPT-Type Flavonoid Derivatives with High Emission Intensity. Materials. 2022; 15(8):2896. https://doi.org/10.3390/ma15082896
Chicago/Turabian StyleYu, Xiangrui, Changjiao Shang, Yunjian Cao, Jingang Cui, and Chaofan Sun. 2022. "A DFT/TD-DFT Study on the ESIPT-Type Flavonoid Derivatives with High Emission Intensity" Materials 15, no. 8: 2896. https://doi.org/10.3390/ma15082896
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.