
Efficient Electrochemical Oxidation of Chloramphenicol by Novel Reduced TiO2 Nanotube Array Anodes: Kinetics, Reaction Parameters, Degradation Pathway and Biotoxicity Forecast
Abstract
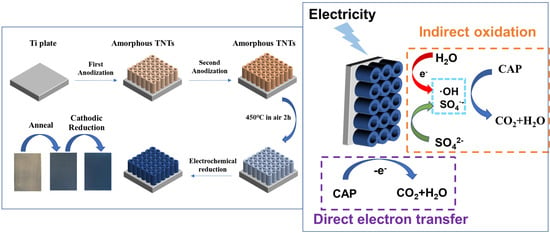
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Experimental Methods
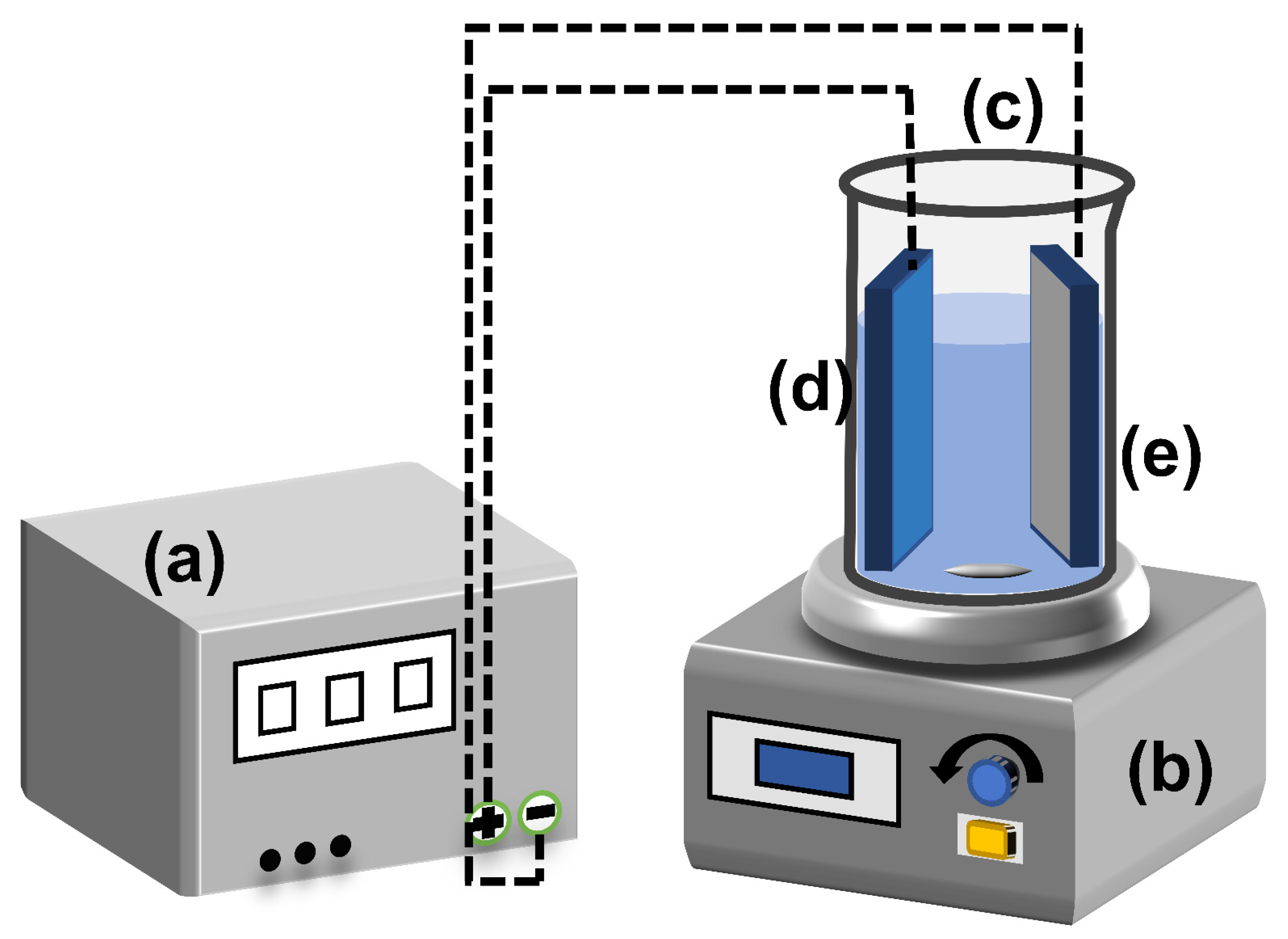
2.2.1. Preparation of TNTs and R-TNTs
2.2.2. Electrocatalytic Degradation of CAP
2.3. Physical and Chemical Property Analysis
3. Results
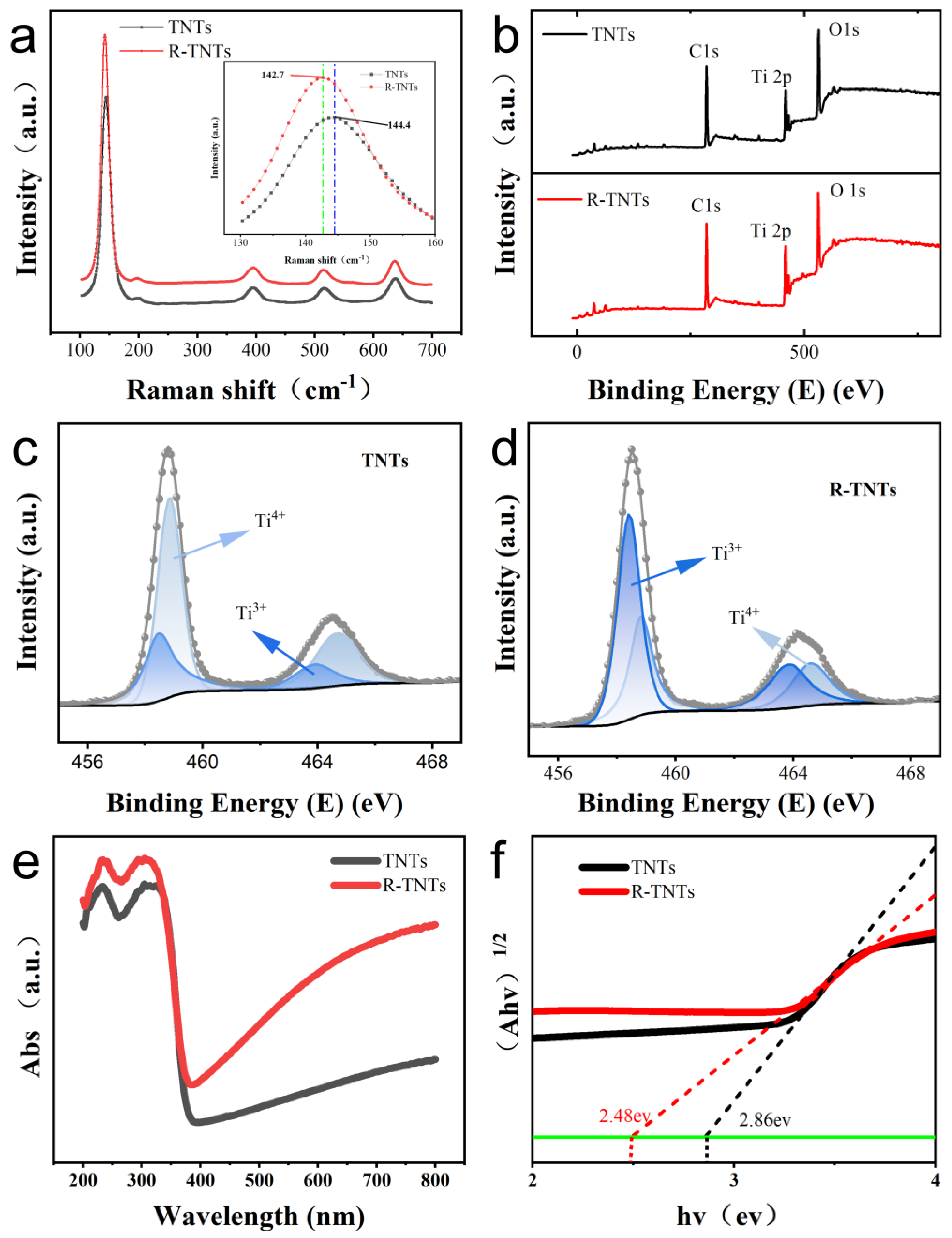
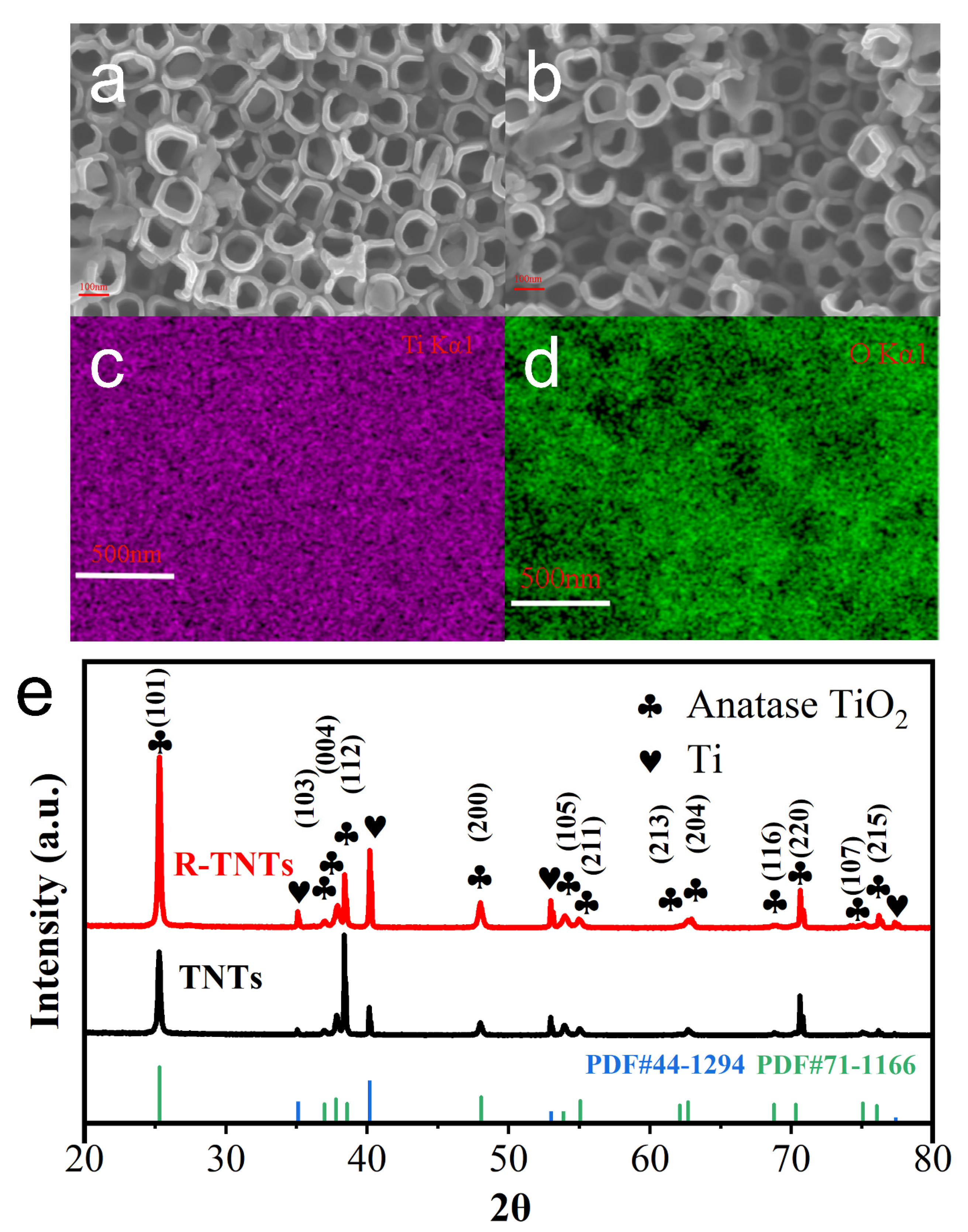
3.1. Electrode Surface Morphology and Structure
3.2. Electrochemical Degradation of CAP
3.2.1. Influence of Anode Materials
3.2.2. Effect of Current Density
3.2.3. Effect of Initial pH
3.2.4. Effect of Initial CAP Concentration
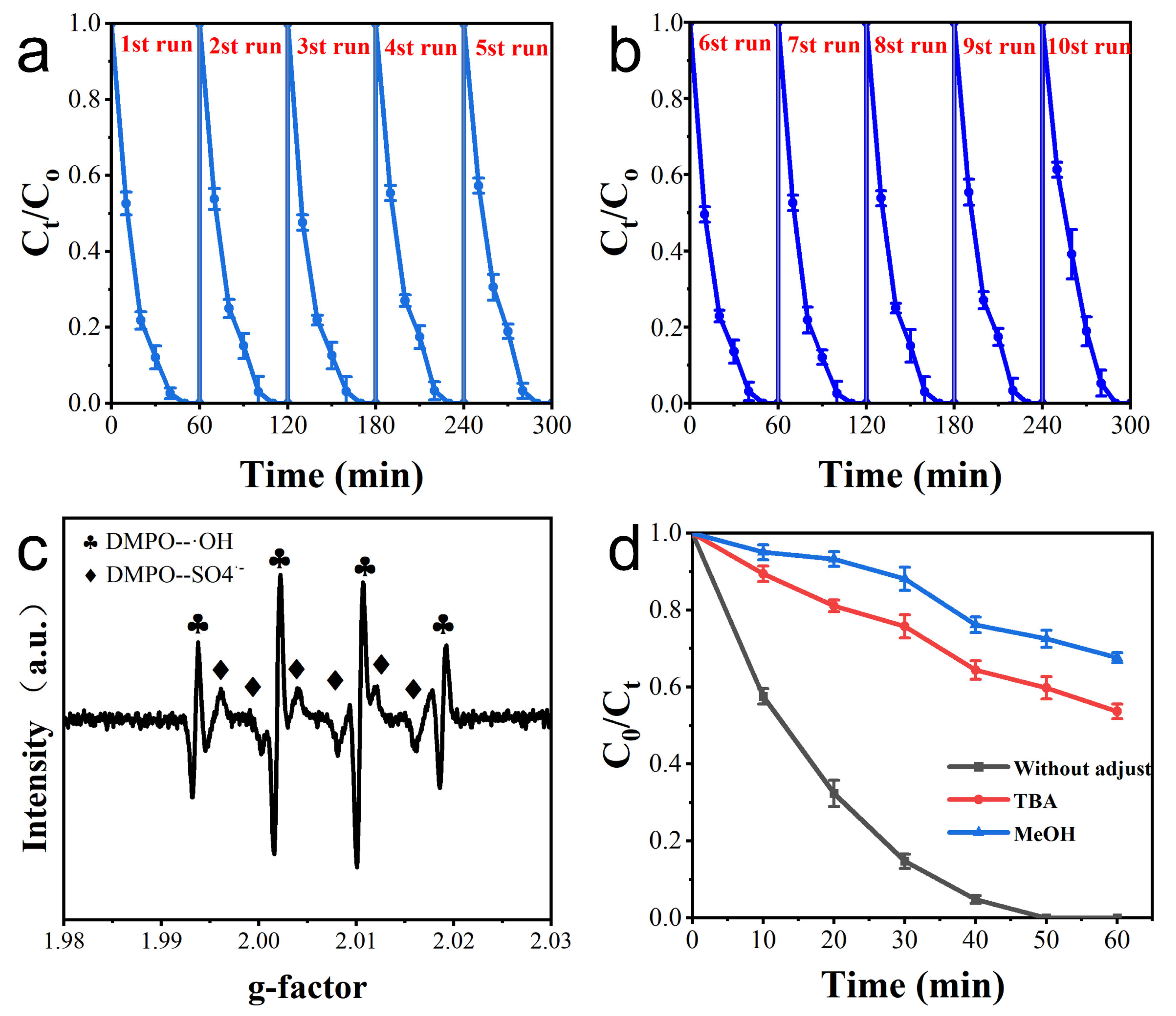
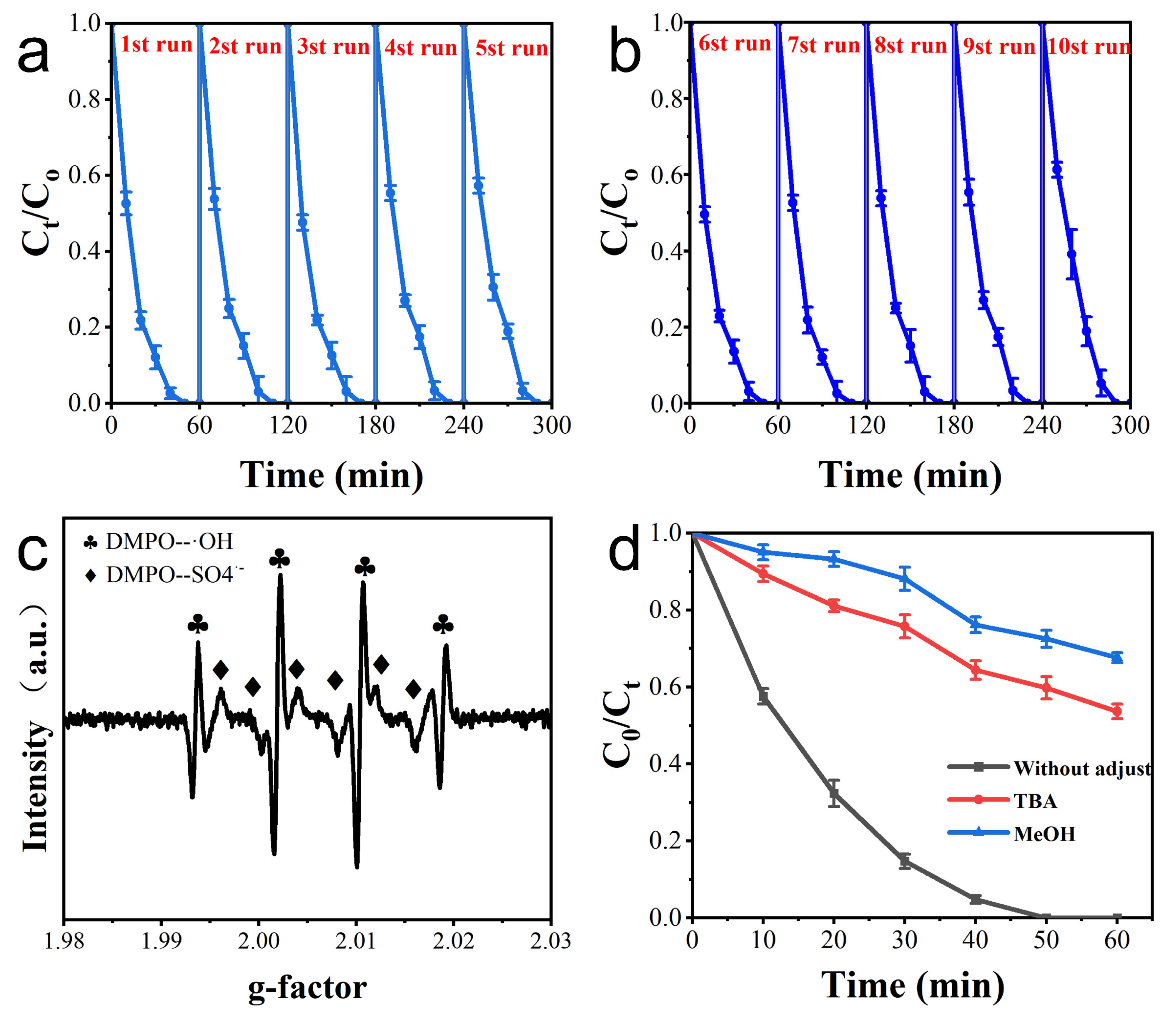
3.3. Electrode Repeatability
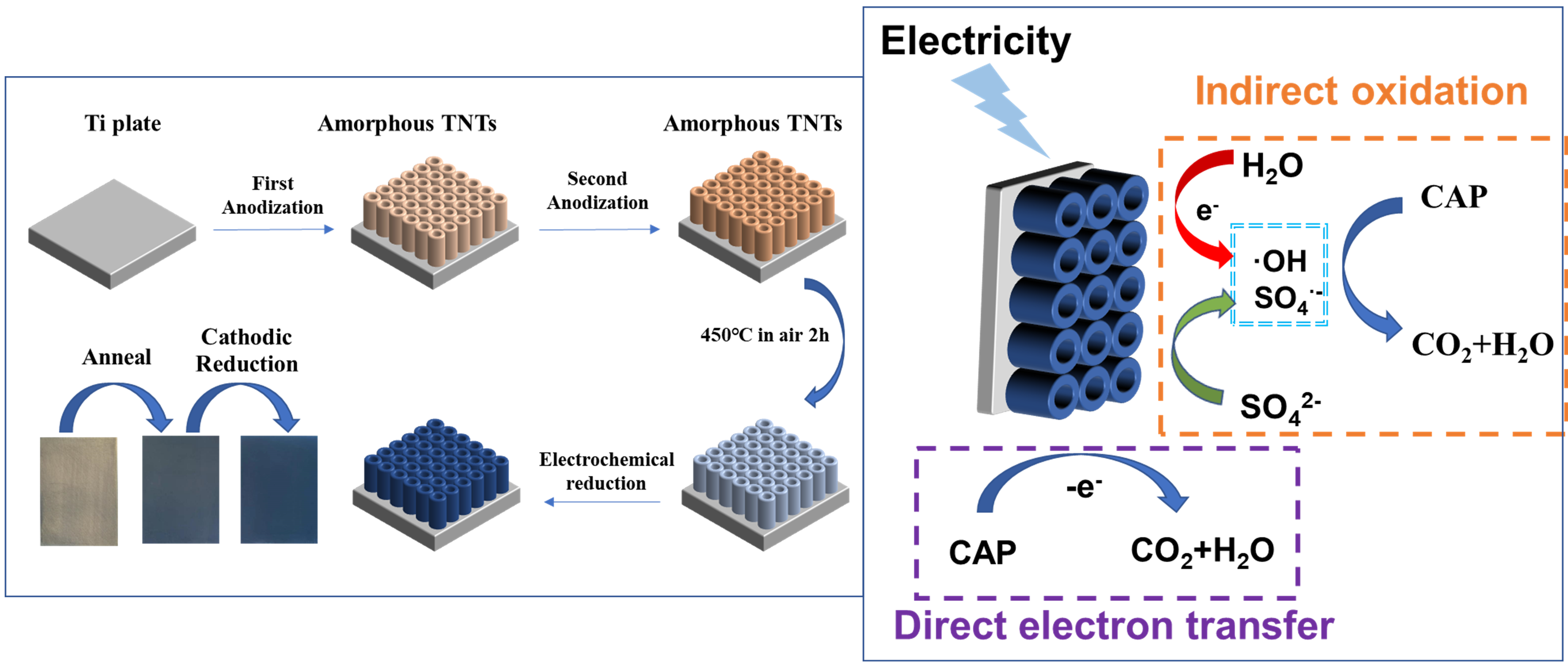
3.4. Assessment of Main Active Species
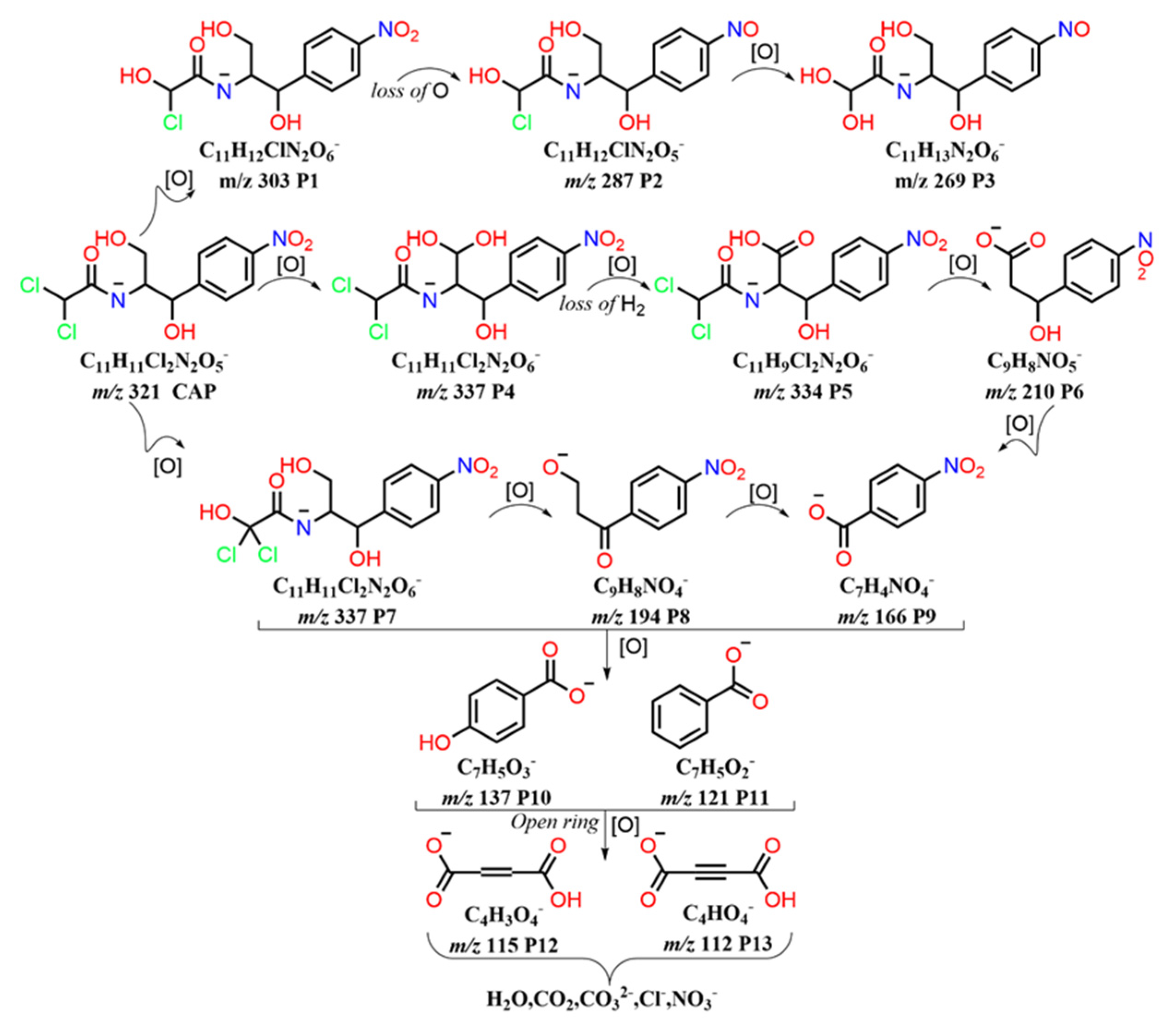
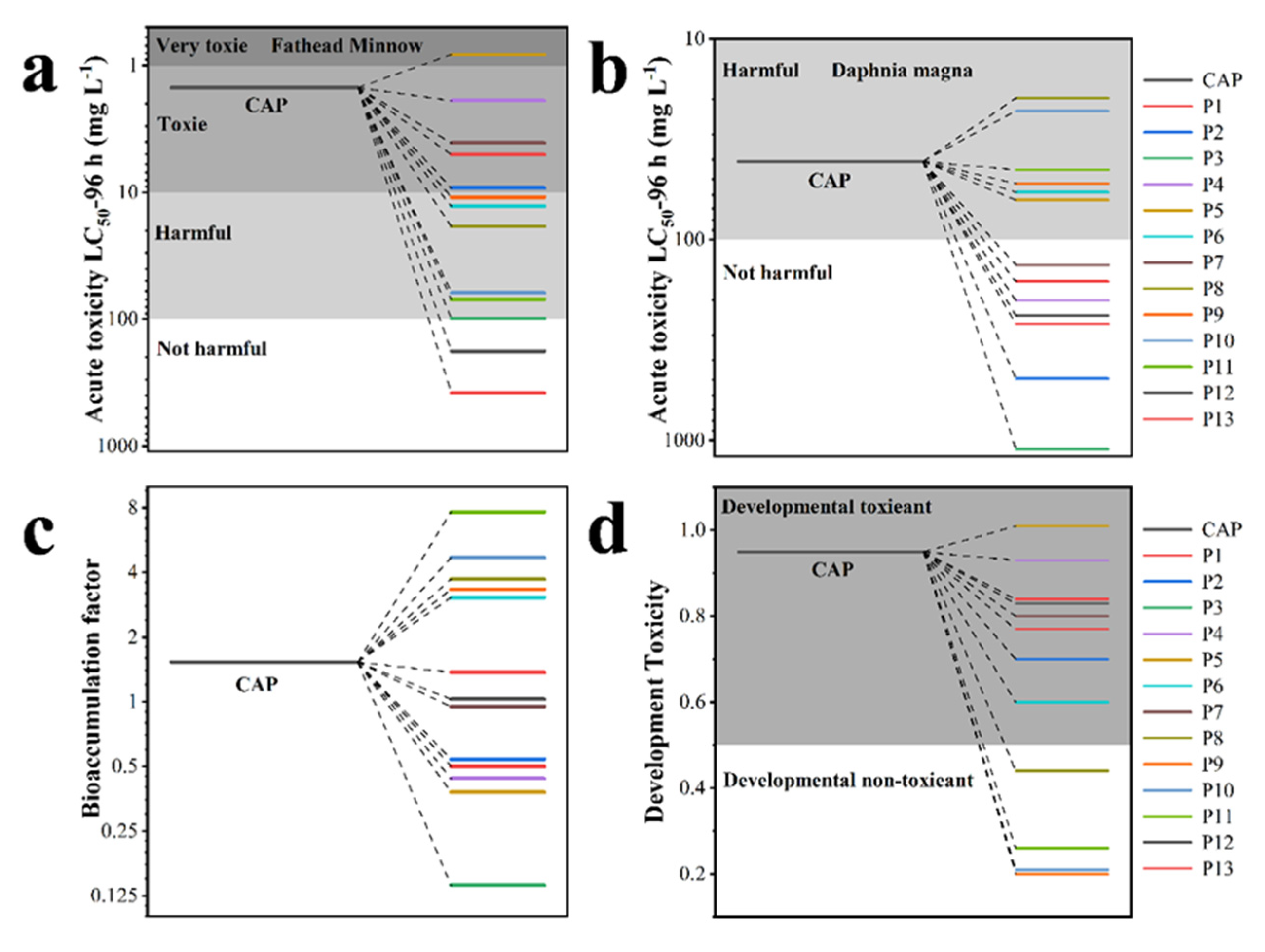
3.5. Electrochemical Degradation Mechanism of CAP and Toxicity Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, B.; Dong, S.; Liu, J.; Liu, L.; Feng, Q.; Tan, N.; Liu, T.; Bo, L.; Wang, L. Identification of Intermediates and Transformation Pathways Derived from Photocatalytic Degradation of Five Antibiotics on ZnIn2S4. Chem. Eng. J. 2016, 304, 826–840. [Google Scholar] [CrossRef]
- Qiao, M.; Ying, G.G.; Singer, A.C.; Zhu, Y.G. Review of Antibiotic Resistance in China and Its Environment. Environ. Int. 2018, 110, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Q.; Ying, G.G.; Pan, C.G.; Liu, Y.S.; Zhao, J.L. Comprehensive Evaluation of Antibiotics Emission and Fate in the River Basins of China: Source Analysis, Multimedia Modeling, and Linkage to Bacterial Resistance. Environ. Sci. Technol. 2015, 49, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Podzelinska, K.; Latimer, R.; Bhattacharya, A.; Vining, L.C.; Zechel, D.L.; Jia, Z. Chloramphenicol Biosynthesis: The Structure of CmlS, a Flavin-dependent Halogenase Showing a Covalent Flavin-aspartate Bond. J. Mol. Biol. 2010, 397, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Feder, J.; Osier, C.; Maderazo, E.G. Chloramphenicol: A Review of Its Use in Clinical Practice. Rev. Infect. Dis. 1981, 3, 479–491. [Google Scholar] [CrossRef]
- Liang, B.; Cheng, H.Y.; Kong, D.Y.; Gao, S.H.; Sun, F.; Cui, D.; Kong, F.Y.; Zhou, A.J.; Liu, W.Z.; Ren, N.Q.; et al. Accelerated Reduction of Chlorinated Nitroaromatic Antibiotic Chloramphenicol by Biocathode. Environ. Sci. Technol. 2013, 47, 5353–5361. [Google Scholar] [CrossRef] [PubMed]
- Kasprzyk-Hordern, B.; Dinsdale, R.M.; Guwy, A.J. The Removal of Pharmaceuticals, Personal Care Products, Endocrine Disruptors and Illicit Drugs During Wastewater Treatment and Its Impact on the Quality of Receiving Waters. Water Res. 2009, 43, 363–380. [Google Scholar] [CrossRef]
- Appavu, B.; Thiripuranthagan, S.; Ranganathan, S.; Erusappan, E.; Kannan, K. BiVO4/N-rGO Nano Composites as Highly Efficient Visible Active Photocatalyst for the Degradation of Dyes and Antibiotics in Eco System. Ecotoxicol. Environ. Saf. 2018, 151, 118–126. [Google Scholar] [CrossRef]
- Tan, C.; Dong, Y.; Fu, D.; Gao, N.; Ma, J.; Liu, X. Chloramphenicol Removal by Zero Valent Iron Activated Peroxymonosulfate System: Kinetics and Mechanism of Radical Generation. Chem. Eng. J. 2018, 334, 1006–1015. [Google Scholar] [CrossRef]
- Ahmed, M.B.; Zhou, J.L.; Ngo, H.H.; Guo, W.; Thomaidis, N.S.; Xu, J. Progress in the Biological and Chemical Treatment Technologies for Emerging Contaminant Removal from Wastewater: A Critical Review. J. Hazard. Mater. 2017, 323, 274–298. [Google Scholar] [CrossRef]
- Ganiyu, S.O.; Vieira Dos Santos, E.; Tossi de Araujo Costa, E.C.; Martinez-Huitle, C.A. Electrochemical Advanced Oxidation Processes (EAOPs) as Alternative Treatment Techniques for Carwash Wastewater Reclamation. Chemosphere 2018, 211, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Titchou, F.E.; Zazou, H.; Afanga, H.; Gaayda, J.E.; Ait Akbour, R.; Nidheesh, P.V.; Hamdani, M. An Overview on the Elimination of Organic Contaminants from Aqueous Systems Using Electrochemical Advanced Oxidation Processes. J. Water Process. Eng. 2021, 41, 102040. [Google Scholar] [CrossRef]
- Canizares, P.; Paz, R.; Saez, C.; Rodrigo, M.A. Costs of the Electrochemical Oxidation of Wastewaters: A Comparison with Ozonation and Fenton Oxidation Processes. J. Environ. Manag. 2009, 90, 410–420. [Google Scholar] [CrossRef]
- Norouzi, R.; Zarei, M.; Khataee, A.; Ebratkhahan, M.; Rostamzadeh, P. Electrochemical Removal of Fluoxetine Via Three Mixed Metal Oxide Anodes and Carbonaceous Cathodes from Contaminated Water. Environ. Res. 2022, 207, 112641. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, Y.; Li, C.; Pierce, R.; Gao, S.; Huang, Q. Degradation of Perfluorooctanesulfonate by Reactive Electrochemical Membrane Composed of Magneli Phase Titanium Suboxide. Environ. Sci. Technol. 2019, 53, 14528–14537. [Google Scholar] [CrossRef]
- Mora-Gomez, J.; Escriba-Jimenez, S.; Carrillo-Abad, J.; Garcia-Gabaldon, M.; Montanes, M.T.; Mestre, S.; Perez-Herranz, V. Study of the Chlorfenvinphos Pesticide Removal Under Different Anodic Materials and Different Reactor Configuration. Chemosphere 2022, 290, 133294. [Google Scholar] [CrossRef]
- Rakhmania; Kamyab, H.; Yuzir, M.A.; Riyadi, F.A.; Ostadrahimi, A.; Khademi, T.; Ghfar, A.A.; Kirpichnikova, I. Electrochemical Oxidation of Palm Oil Mill Effluent Using Platinum as Anode: Optimization Using Response Surface Methodology. Environ. Res. 2022, 214, 113993. [Google Scholar] [CrossRef]
- Radjenovic, J.; Sedlak, D.L. Challenges and Opportunities for Electrochemical Processes as Next-Generation Technologies for the Treatment of Contaminated Water. Environ. Sci. Technol. 2015, 49, 11292–11302. [Google Scholar] [CrossRef]
- Yang, Y.; Kao, L.C.; Liu, Y.; Sun, K.; Yu, H.; Guo, J.; Liou, S.Y.H.; Hoffmann, M.R. Cobalt-doped Black TiO2 Nanotube Array as A Stable Anode for Oxygen Evolution and Electrochemical Wastewater Treatment. ACS Catal. 2018, 8, 4278–4287. [Google Scholar] [CrossRef]
- Fujishima, A.H.K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Rajaraman, T.S.; Parikh, S.P.; Gandhi, V.G. Black TiO2: A Review of Its Properties and Conflicting Trends. Chem. Eng. J. 2020, 389, 123918. [Google Scholar] [CrossRef]
- Roy, P.; Berger, S.; Schmuki, P. TiO2 Nanotubes: Synthesis and Applications. Angew. Chem. Int. Ed. 2011, 50, 2904–2939. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.Y.; Roy, P.; Paramasivam, I.; Schmuki, P. Voltage-Induced Payload Release and Wettability Control on TiO2 and TiO2 Nanotubes. Angew. Chem. Int. Ed. 2010, 49, 351–354. [Google Scholar] [CrossRef]
- Galstyan, V.; Macak, J.M.; Djenizian, T. Anodic TiO2 Nanotubes: A Promising Material for Energy Conversion and Storage. Appl. Mater. 2022, 29, 101613. [Google Scholar] [CrossRef]
- Li, J.; Lu, N.; Quan, X.; Chen, S. Facile Method for Fabricating Boron-Doped TiO2 Nanotube Array with Enhanced Photoelectrocatalytic Properties. J. Ind. Eng. Chem. 2008, 47, 3804–3808. [Google Scholar] [CrossRef]
- Hu, Z.; Cai, J.; Song, G.; Tian, Y.; Zhou, M. Anodic Oxidation of Organic Pollutants: Anode Fabrication, Process Hybrid and Environmental Applications. Curr. Opin. Electrochem. 2021, 26, 100659. [Google Scholar] [CrossRef]
- Zaky, A.M.; Chaplin, B.P. Mechanism of P-Substituted Phenol Oxidation at A Ti4O7 Reactive Electrochemical Membrane. Environ. Sci. Technol. 2014, 48, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zhu, X.; Xu, Z.; Zhong, X.; Gui, Q.; Song, Y.; Zhang, S.; Chen, X.; Li, D. Facile Method to Enhance the Adhesion of TiO2 Nanotube Arrays to Ti Substrate. ACS Appl. Mater. 2014, 6, 8001–8005. [Google Scholar] [CrossRef]
- Cai, J.; Zhou, M.; Pan, Y.; Du, X.; Lu, X. Extremely Efficient Electrochemical Degradation of Organic Pollutants with Co-Generation of Hydroxyl and Sulfate Radicals on Blue-TiO2 Nanotubes Anode. Appl. Catal. B 2019, 257, 117902. [Google Scholar] [CrossRef]
- Chang, X.; Thind, S.S.; Chen, A. Electrocatalytic Enhancement of Salicylic Acid Oxidation at Electrochemically Reduced TiO2 Nanotubes. ACS Catal. 2014, 4, 2616–2622. [Google Scholar] [CrossRef]
- Han, S.C.; Doh, J.M.; Yoon, J.K.; Kim, G.H.; Byun, J.Y.; Han, S.H.; Hong, K.T.; Kwun, S.I. Highly Ordered Self-Organized TiO2 Nanotube Arrays Prepared by A Multi-Step Anodic Oxidation Process. Met. Mater. Int. 2009, 15, 493–499. [Google Scholar] [CrossRef]
- Xu, X.; Cai, J.; Zhou, M.; Du, X.; Zhang, Y. Photoelectrochemical Degradation of 2,4-Dichlorophenoxyacetic Acid Using Electrochemically Self-Doped Blue TiO2 Nanotube Arrays with Formic Acid as Electrolyte. J. Hazard. 2020, 382, 121096. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhao, G.; Zhao, K.; Tong, X.; Tang, Y. Direct Electrochemistry of Hemoglobin at Vertically-aligned Self-doping TiO2 Nanotubes: A Mediator-Free and Biomolecule-Substantive Electrochemical Interface. Electrochem. Commun. 2009, 11, 1397–1400. [Google Scholar] [CrossRef]
- Zhu, W.-D.; Wang, C.-W.; Chen, J.-B.; Li, Y.; Wang, J. Enhanced Field Emission from Ti3+ Self-doped TiO2 Nanotube Arrays Synthesized by A Facile Cathodic Reduction Process. Appl. Surf. Sci. 2014, 301, 525–529. [Google Scholar] [CrossRef]
- Zeng, W.; Liang, H.; Zhang, H.; Luo, X.; Lin, D.; Li, G. Efficient Electrochemical Oxidation of Sulfamethoxazole by A Novel Reduced TiO2 Nanotube Arrays-based Flow-through Electrocatalytic Membrane. Sep. Purif. 2022, 289, 120720. [Google Scholar] [CrossRef]
- Yang, Y.; Hoffmann, M.R. Synthesis and Stabilization of Blue-black TiO2 Nanotube Arrays for Electrochemical Oxidant Generation and Wastewater Treatment. Environ. Sci. Technol. 2016, 50, 11888–11894. [Google Scholar] [CrossRef]
- Macak, J.M.; Tsuchiya, H.; Ghicov, A.; Hahn, R.; Bauer, S.; Schmuki, P. TiO2 Nanotubes: Self-organized Electrochemical Formation, Properties and Applications. Curr. Opin Solid State Mater. Sci. 2007, 11, 3–18. [Google Scholar] [CrossRef]
- Liao, W.; Yang, J.; Zhou, H.; Murugananthan, M.; Zhang, Y. Electrochemically Self-Doped TiO2 Nanotube Arrays for Efficient Visible Light Photoelectrocatalytic Degradation of Contaminants. Electrochim. Acta 2014, 136, 310–317. [Google Scholar] [CrossRef]
- Feng, X.; Wang, P.; Hou, J.; Qian, J.; Ao, Y.; Wang, C. Significantly Enhanced Visible Light Photocatalytic Efficiency of Phosphorus Doped TiO2 with Surface Oxygen Vacancies for Ciprofloxacin Degradation: Synergistic Effect and Intermediates Analysis. J. Hazard. 2018, 351, 196–205. [Google Scholar] [CrossRef]
- Titchou, F.E.; Zazou, H.; Afanga, H.; Jamila, E.G.; Ait Akbour, R.; Hamdani, M.; Oturan, M.A. Comparative Study of the Removal of Direct Red 23 by Anodic Oxidation, Electro-fenton, Photo-anodic Oxidation and Photoelectro-fenton in Chloride and Sulfate Media. Environ. Res. 2022, 204, 112353. [Google Scholar] [CrossRef]
- Naldoni, A.; Allieta, M.; Santangelo, S.; Marelli, M.; Fabbri, F.; Cappelli, S.; Bianchi, C.L.; Psaro, R.; Dal Santo, V. Effect of Nature and Location of Defects on Bandgap Narrowing in Black TiO2 Nanoparticles. J. Am. Chem. Soc. 2012, 134, 7600–7603. [Google Scholar] [CrossRef] [PubMed]
- Palma-Goyes, R.E.; Guzman-Duque, F.L.; Penuela, G.; Gonzalez, I.; Nava, J.L.; Torres-Palma, R.A. Electrochemical Degradation of Crystal Violet with BDD Electrodes: Effect of Electrochemical Parameters and Identification of Organic By-Products. Chemosphere 2010, 81, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Meng, X.; Sun, P.; Niu, J.; Jiang, W.; Bottomley, L.; Li, D.; Chen, Y.; Crittenden, J. Electrochemical Oxidation of Ofloxacin Using A TiO2-based SnO2-Sb/Polytetrafluoroethylene Resin-PbO2 Electrode: Reaction Kinetics and Mass Transfer Impact. Appl. Catal. B 2017, 203, 515–525. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, C.; Lu, J.; Hu, C.; Peng, S.; Chen, T. Electro-catalytic Degradation of Bisphenol A with Modified Co3O4/β-PbO2/Ti Electrode. Electrochim. Acta 2014, 118, 169–175. [Google Scholar] [CrossRef]
- Qian, L.; Yan, S.; Yong, X.; Selvaraj, M.; Ghramh, H.A.; Assiri, M.A.; Zhang, X.; Awasthi, M.K.; Zhou, J. Effective Degradation of Chloramphenicol in Wastewater by Activated Peroxymonosulfate with Fe-rich Porous Biochar Derived from Petrochemical Sludge. Chemosphere 2023, 310, 136839. [Google Scholar] [CrossRef]
- Brienza, M.; Garcia-Segura, S. Electrochemical oxidation of Fipronil Pesticide is Effective under Environmental Relevant Concentrations. Chemosphere 2022, 307, 135974. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH/·O− in Aqueous Solution). J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Xue, J.; Chen, L.; Wang, H. Degradation Mechanism of Alizarin Red in Hybrid Gas–liquid Phase Dielectric Barrier Discharge Plasmas: Experimental and Theoretical Examination. Chem. Eng. J. 2008, 138, 120–127. [Google Scholar] [CrossRef]
- Lou, J.; Lu, N.; Li, J.; Wang, T.; Wu, Y. Remediation of Chloramphenicol-contaminated Soil by Atmospheric Pressure Dielectric Barrier Discharge. Chem. Eng. J. 2012, 180, 99–105. [Google Scholar] [CrossRef]
- Marson, E.O.; Paniagua, C.E.S.; Costa-Serge, N.M.; Sousa, R.M.F.; Silva, G.D.; Becker, R.W.; Sirtori, C.; Starling, M.C.V.M.; Carvalho, S.R.; Trovó, A.G. Chemical and Toxicological Evaluation along with Unprecedented Transformation Products during Photolysis and Heterogeneous Photocatalysis of Chloramphenicol in Different Aqueous Matrices. Environ. Sci. Pollut. Res. 2020, 28, 23582–23594. [Google Scholar] [CrossRef]
- Amildon Ricardo, I.; Paniagua, C.E.S.; Paiva, V.A.B.; Gonçalves, B.R.; Sousa, R.M.F.; Machado, A.E.H.; Trovó, A.G. Degradation and Initial Mechanism Pathway of Chloramphenicol by photo-Fenton Process at Circumneutral pH. Chem. Eng. J. 2018, 339, 531–538. [Google Scholar] [CrossRef]
- Yi, X.-H.; Ji, H.; Wang, C.-C.; Li, Y.; Li, Y.-H.; Zhao, C.; Wang, A.; Fu, H.; Wang, P.; Zhao, X.; et al. Photocatalysis-activated SR-AOP over PDINH/MIL-88A(Fe) Composites for Boosted Chloroquine Phosphate Degradation: Performance, Mechanism, Pathway and DFT Calculations. Appl. Catal. B 2021, 293, 120229. [Google Scholar] [CrossRef]
- Taveira, L.V.; Macák, J.M.; Tsuchiya, H.; Dick, L.F.P.; Schmuki, P. Initiation and Growth of Self-organized TiO2 Nanotubes Anodically Formed in NH4F/(NH4)2SO4 Electrolytes. J. Electrochem. Soc. 2005, 152, B405. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Chu, G.; Gao, G.; Li, F.; Ren, Y.; Ding, Y.; Gu, Y.; Jiang, W.; Zhang, X. Efficient Electrochemical Oxidation of Chloramphenicol by Novel Reduced TiO2 Nanotube Array Anodes: Kinetics, Reaction Parameters, Degradation Pathway and Biotoxicity Forecast. Materials 2023, 16, 3971. https://doi.org/10.3390/ma16113971
Wang P, Chu G, Gao G, Li F, Ren Y, Ding Y, Gu Y, Jiang W, Zhang X. Efficient Electrochemical Oxidation of Chloramphenicol by Novel Reduced TiO2 Nanotube Array Anodes: Kinetics, Reaction Parameters, Degradation Pathway and Biotoxicity Forecast. Materials. 2023; 16(11):3971. https://doi.org/10.3390/ma16113971
Chicago/Turabian StyleWang, Pengqi, Guangyi Chu, Guangfei Gao, Fengchun Li, Yi Ren, Yue Ding, Yawei Gu, Wenqiang Jiang, and Xuan Zhang. 2023. "Efficient Electrochemical Oxidation of Chloramphenicol by Novel Reduced TiO2 Nanotube Array Anodes: Kinetics, Reaction Parameters, Degradation Pathway and Biotoxicity Forecast" Materials 16, no. 11: 3971. https://doi.org/10.3390/ma16113971