Recent Progress in Advanced Materials for Lithium Ion Batteries

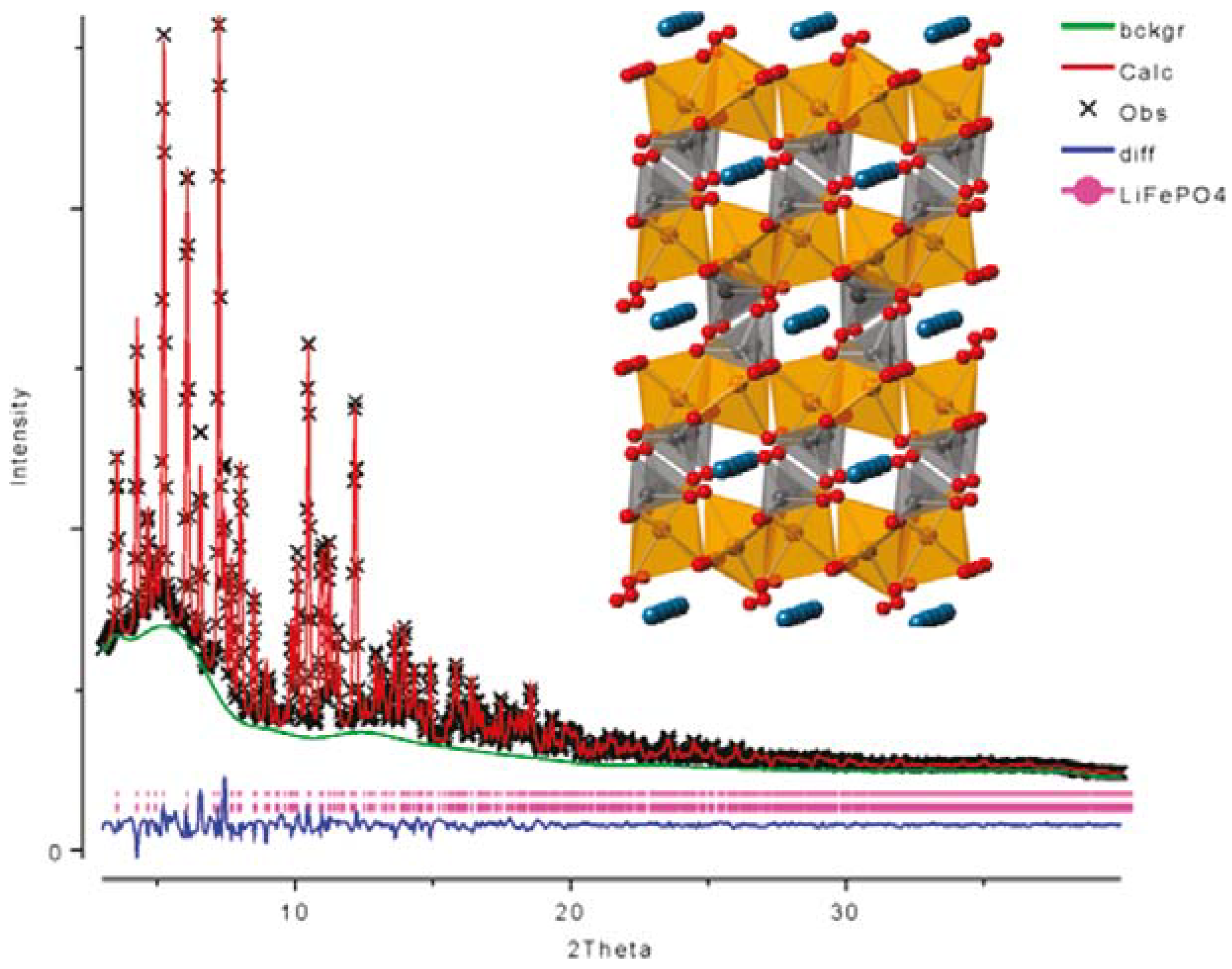{kind=link}
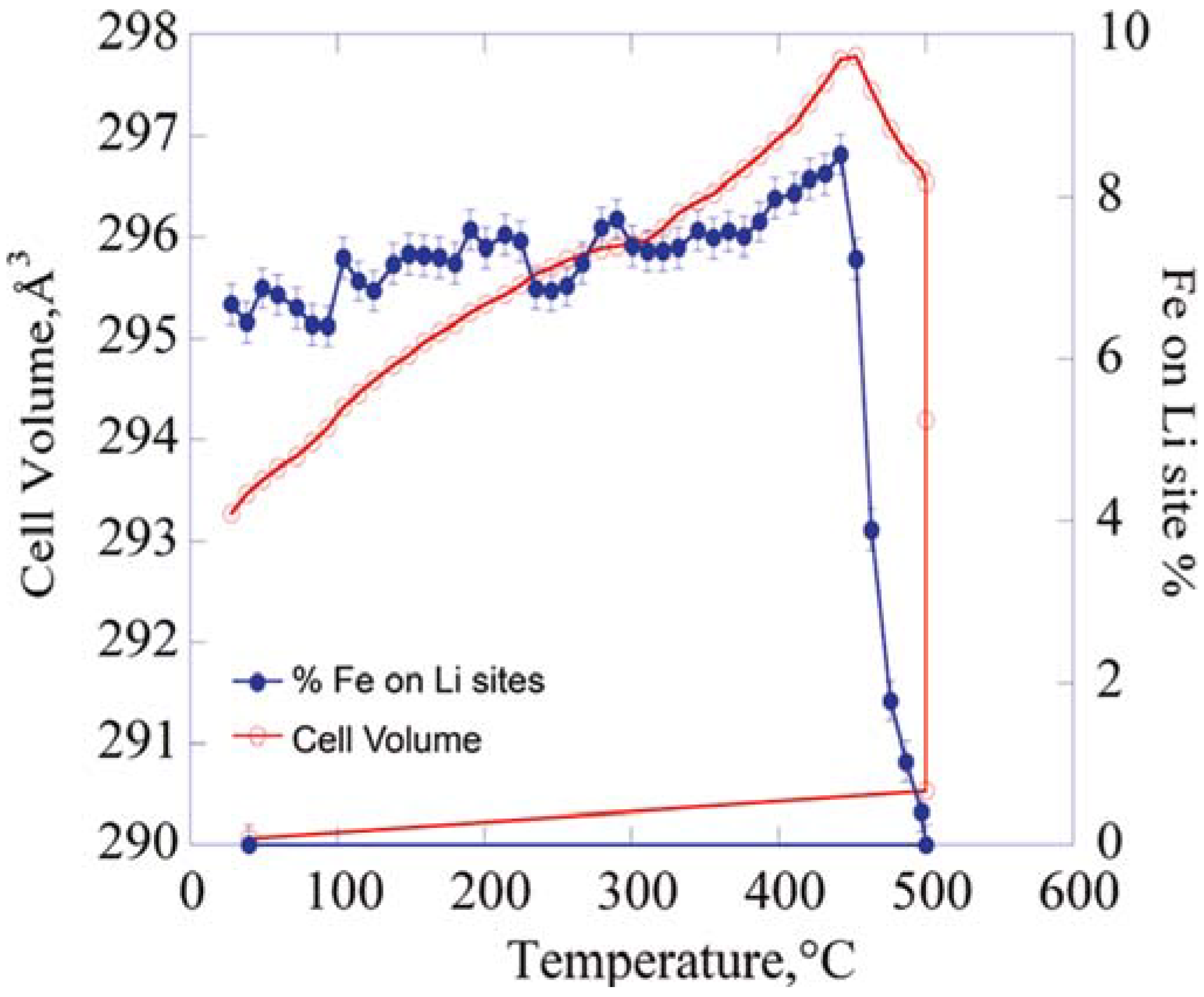{kind=link}
{kind=link}
{kind=link}
{kind=link}
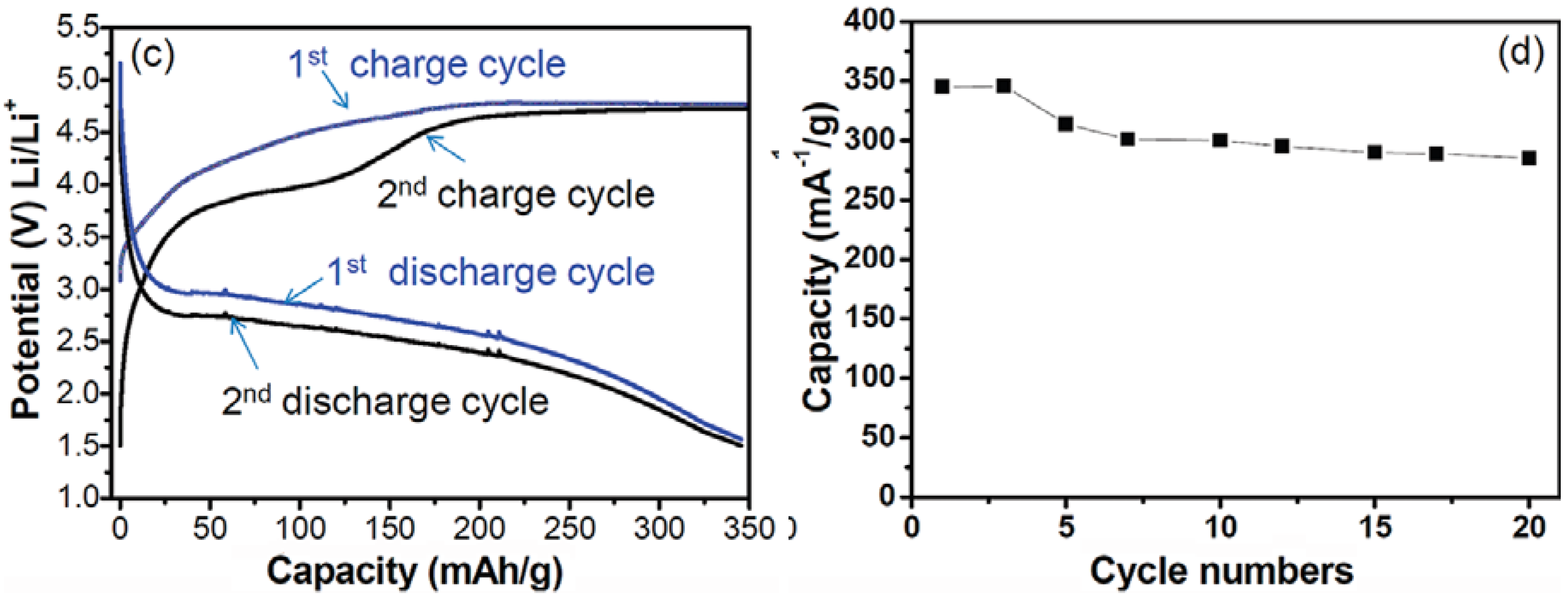{kind=link}
{kind=link}
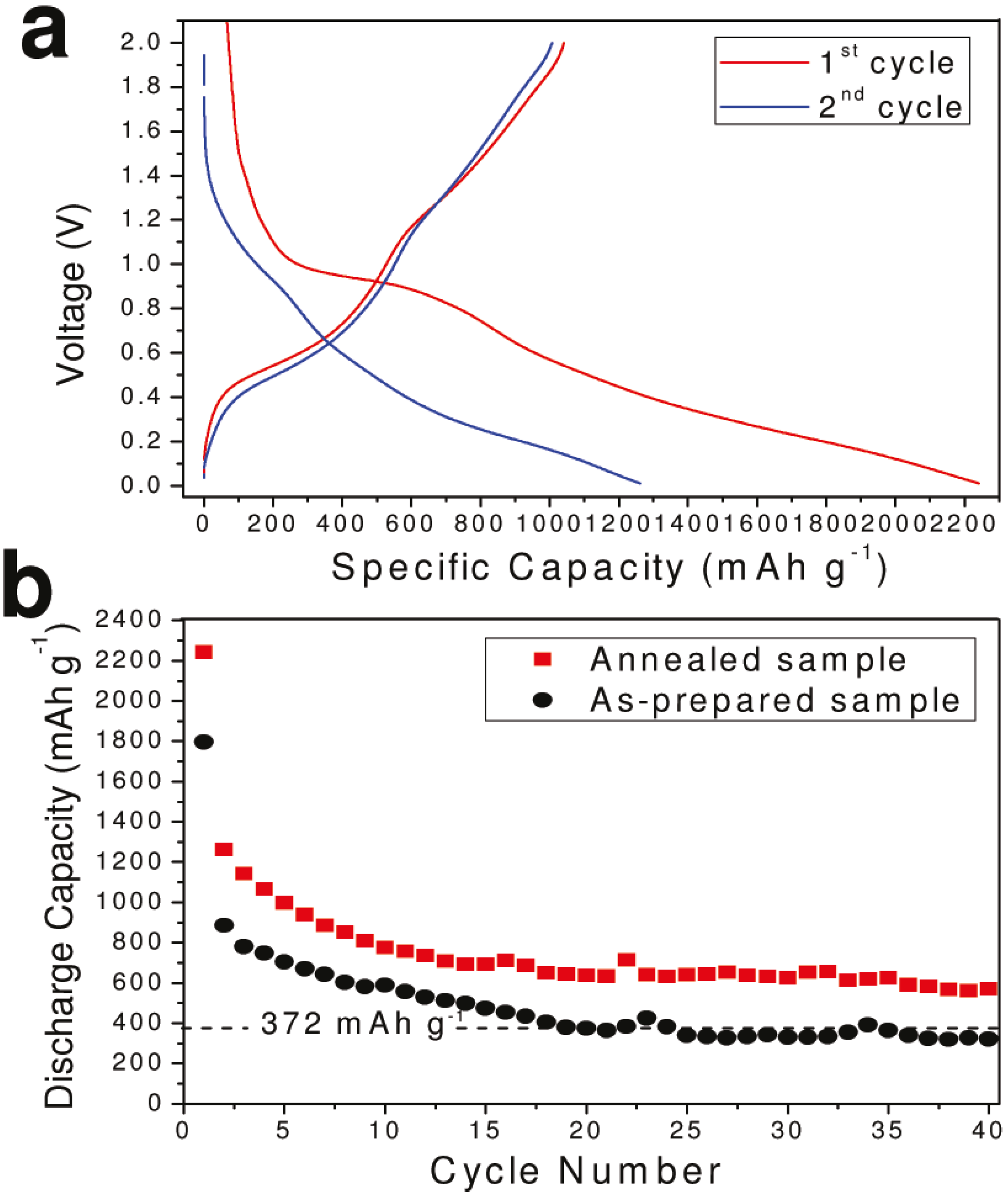{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cathodes
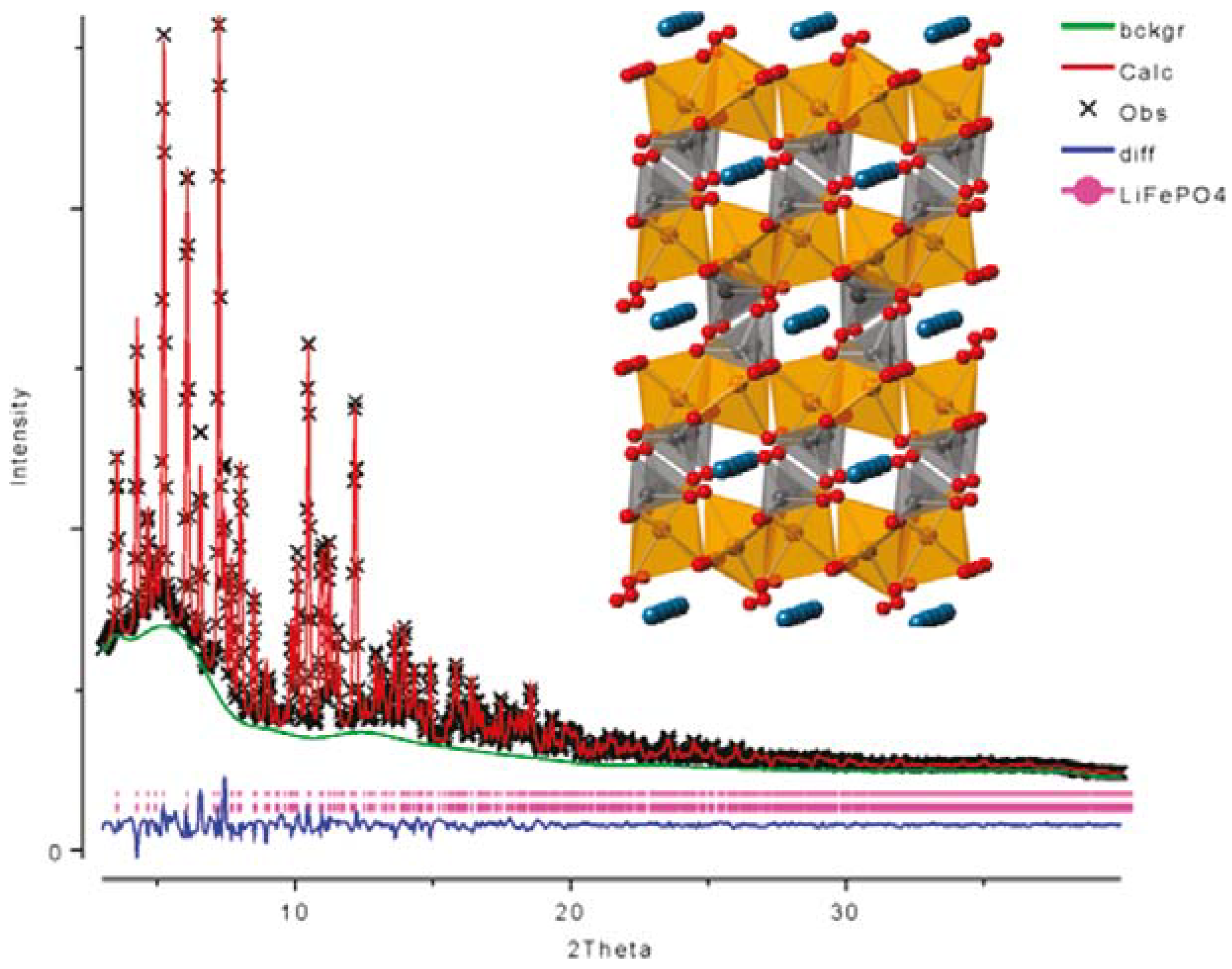
2.1. Olivine-Structured LiMPO4 (M = Fe, Mn, Co, Ni)
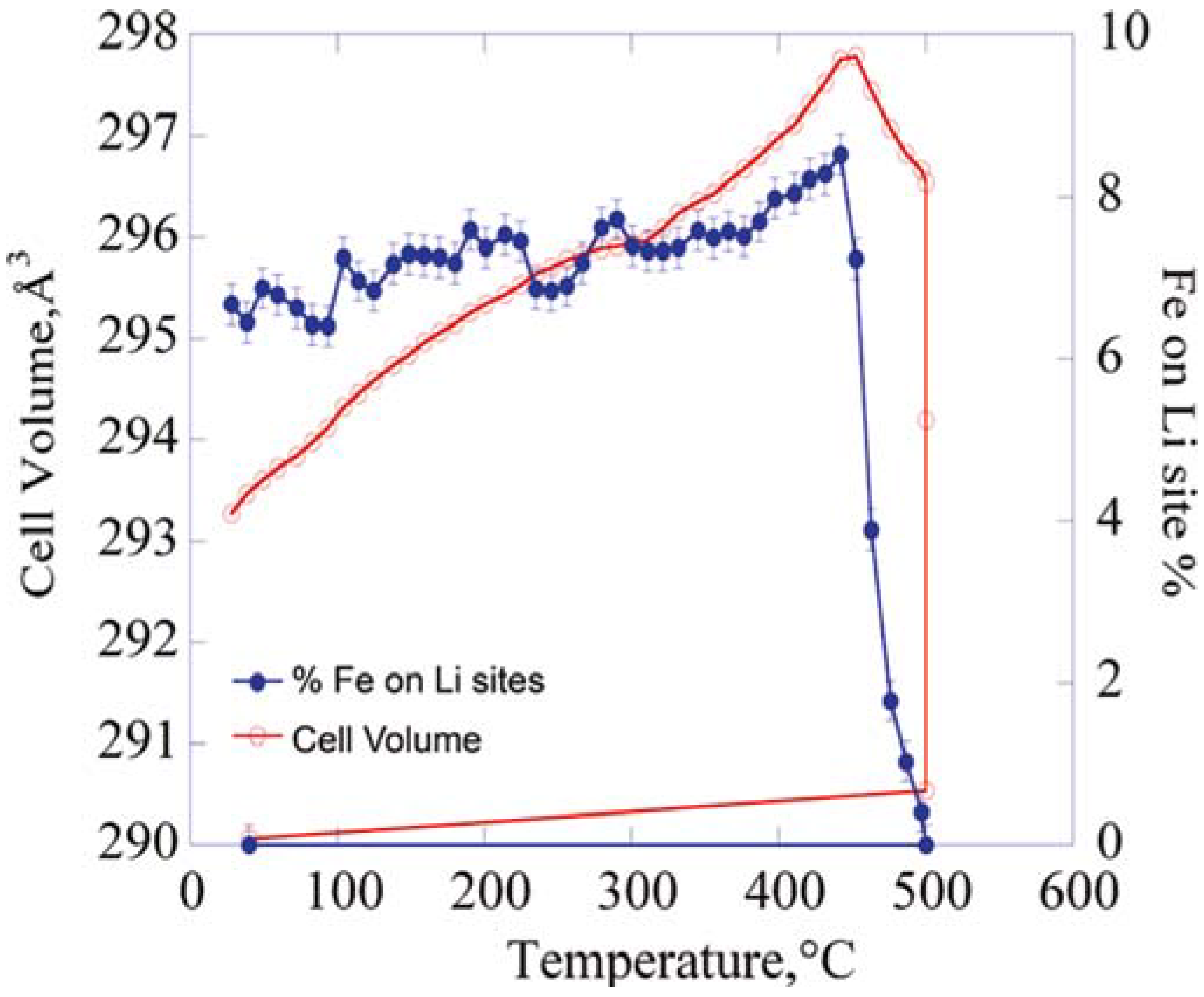
2.2. Orthosilicates Li2MSiO4 (M = Fe, Mn, Co)
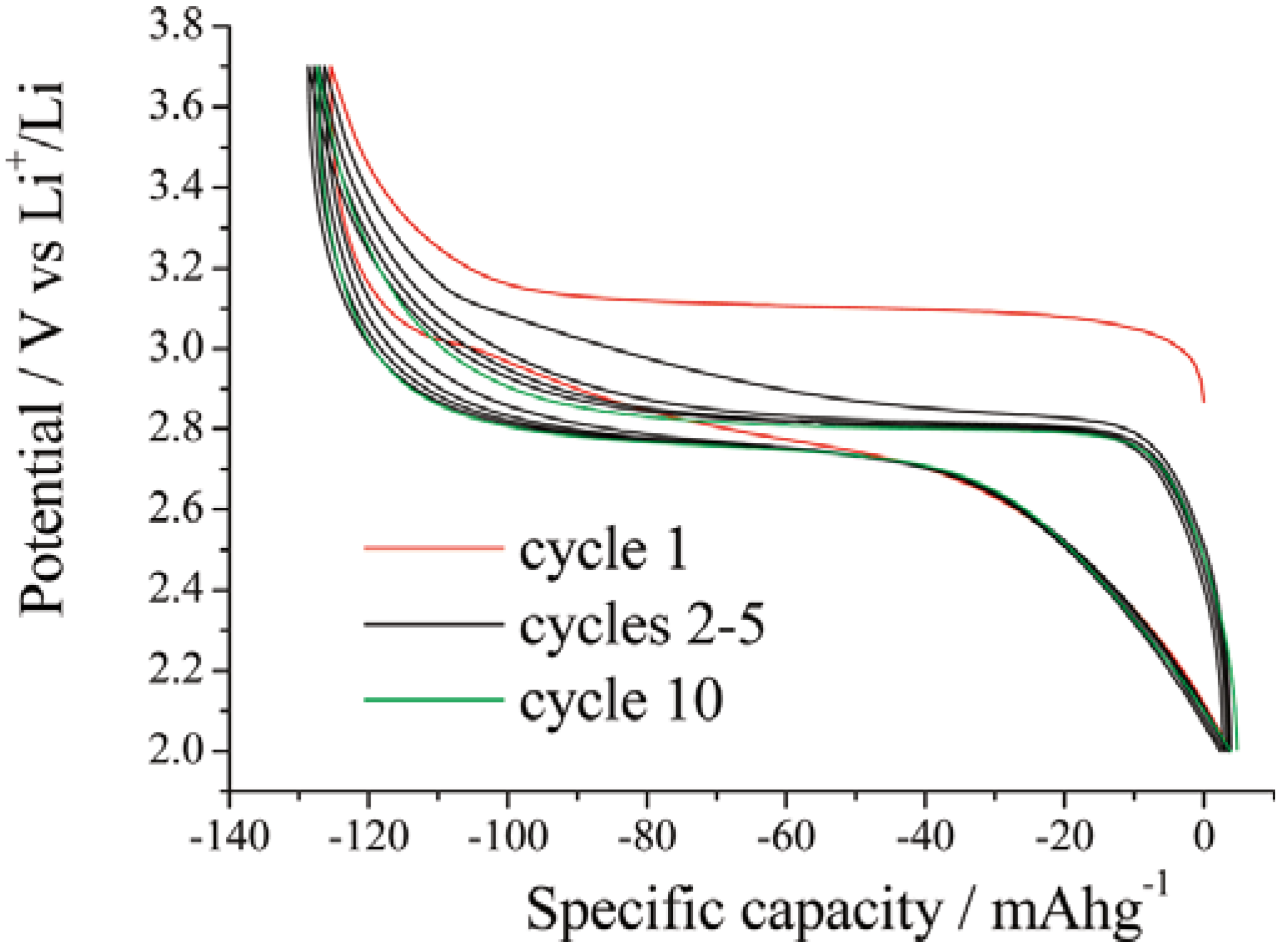

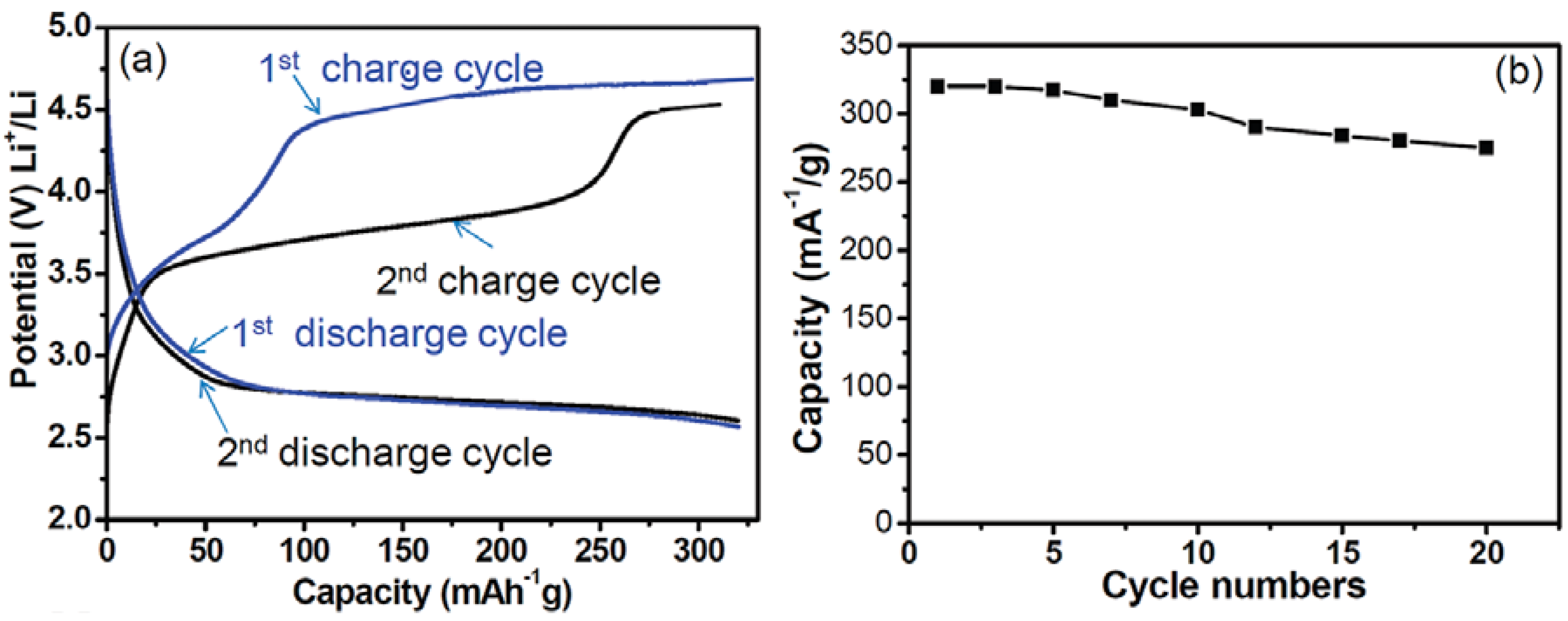

3. Anodes
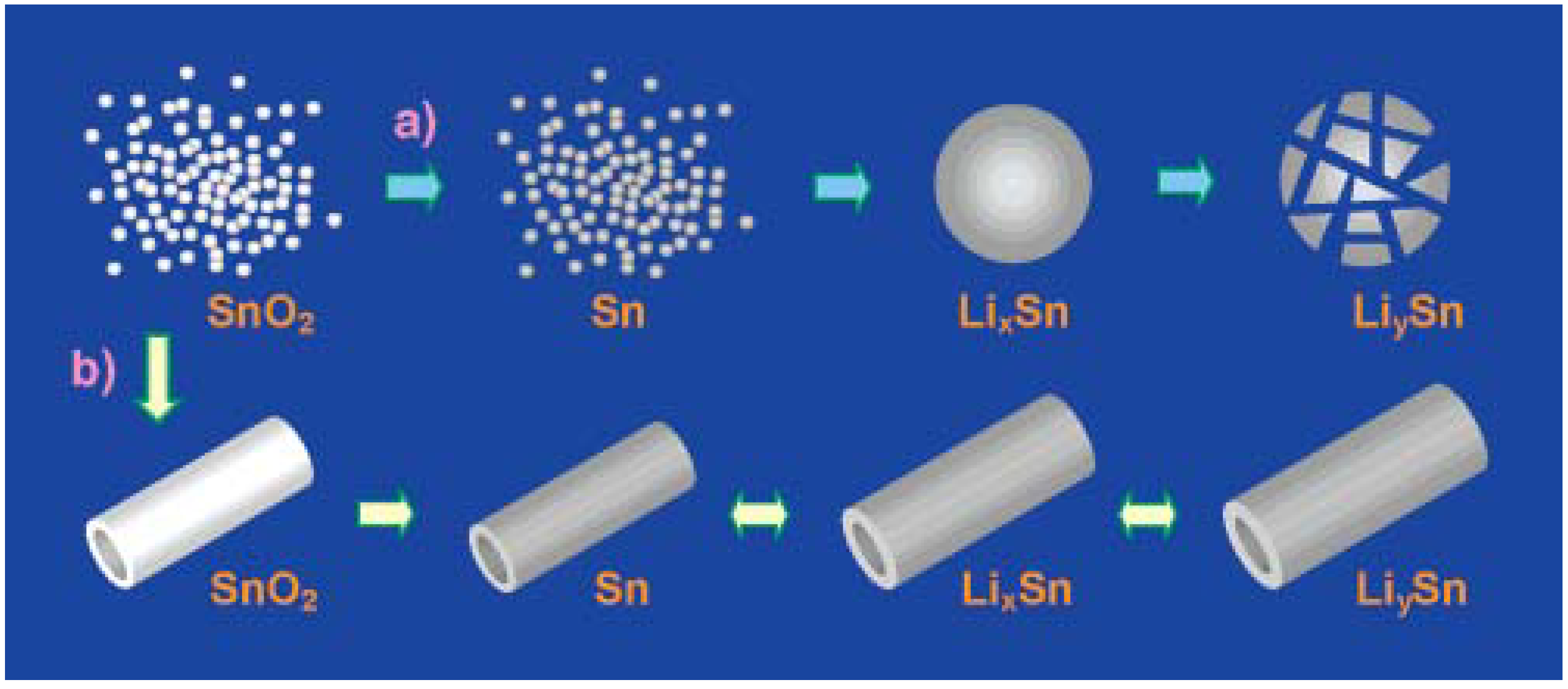
3.1. Tin-Based Materials as Advanced Anode Materials for Lithium Ion Batteries
3.1.1. Pure Tin Anode
3.1.2. Tin/Carbon Composite
3.1.3. Tin–(M)–Carbon (M = Co, Fe, Ti)
3.1.4. Tin Oxide Materials
3.1.4.1. Tin Oxide Nanoparticles
3.1.4.2. Tin Oxide Hollow Structures

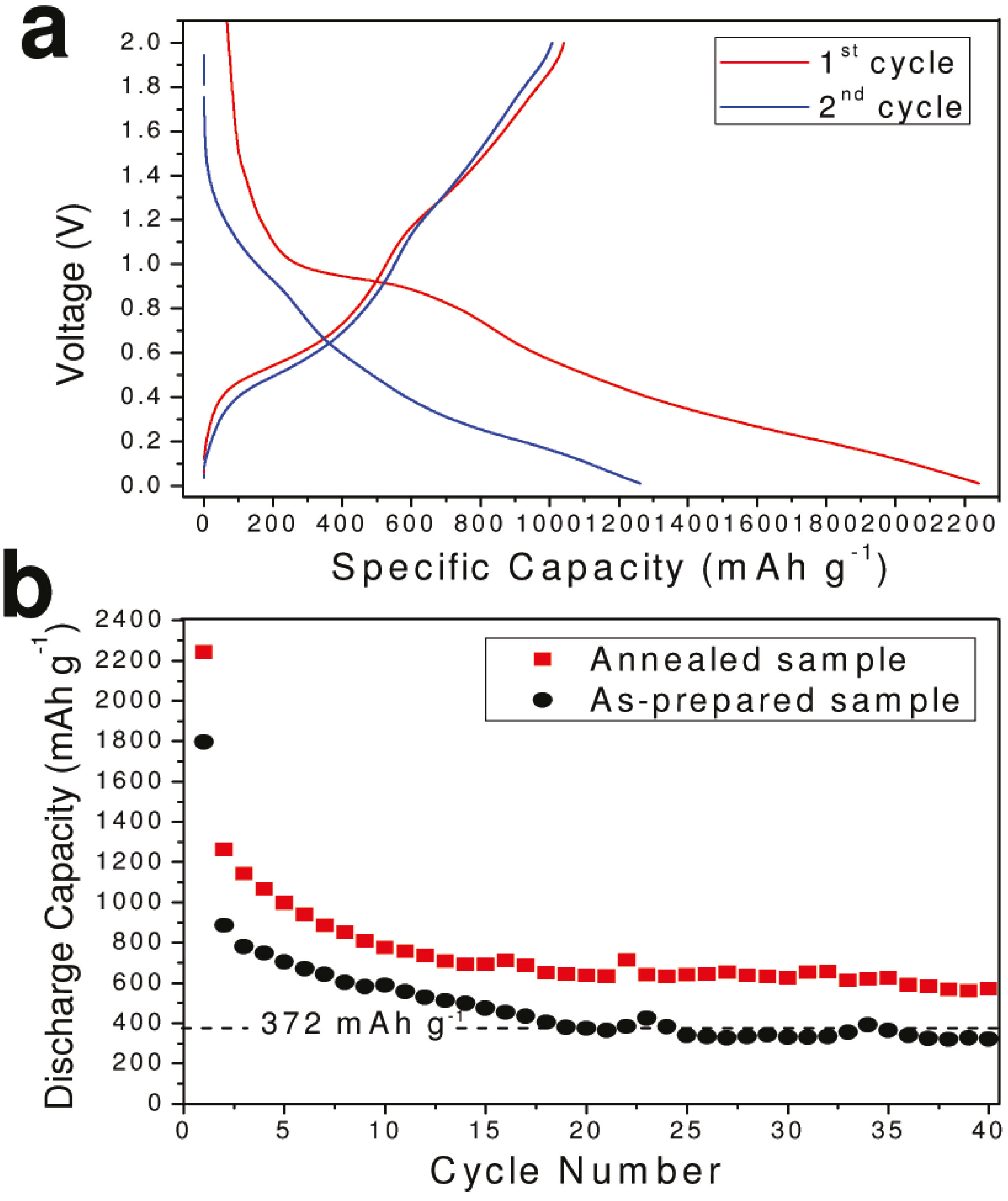
3.1.4.3. Tin Oxide Nanotubes

3.1.4.4. Tin Oxide Nanowires
3.1.4.5. Tin Oxide Nanosheets
3.1.4.6. Tin Oxide/Carbon Composite
3.1.4.7. In situ Observation of the Electrochemical Lithiation of Tin Oxide Electrode
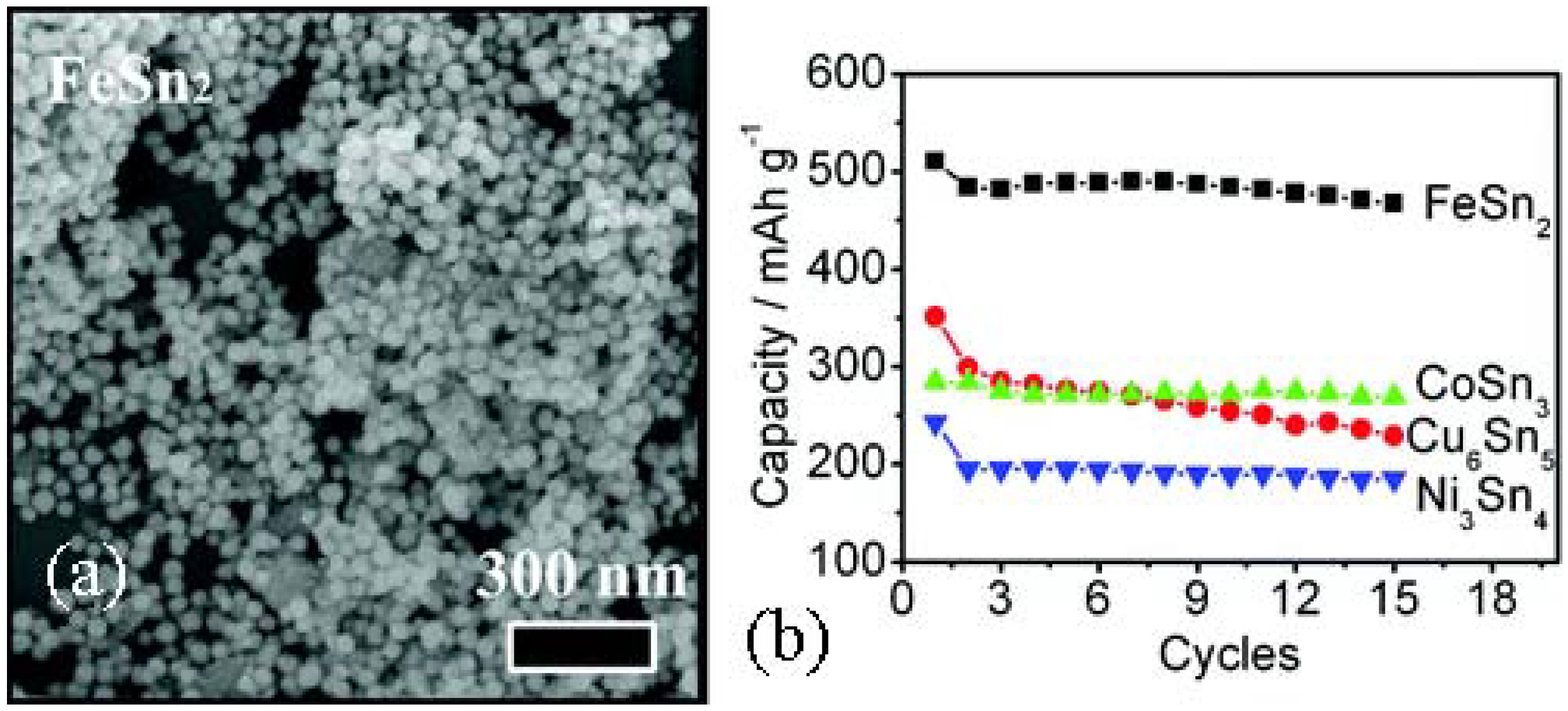
3.1.5. Tin-Based Intermetallic Anode Materials

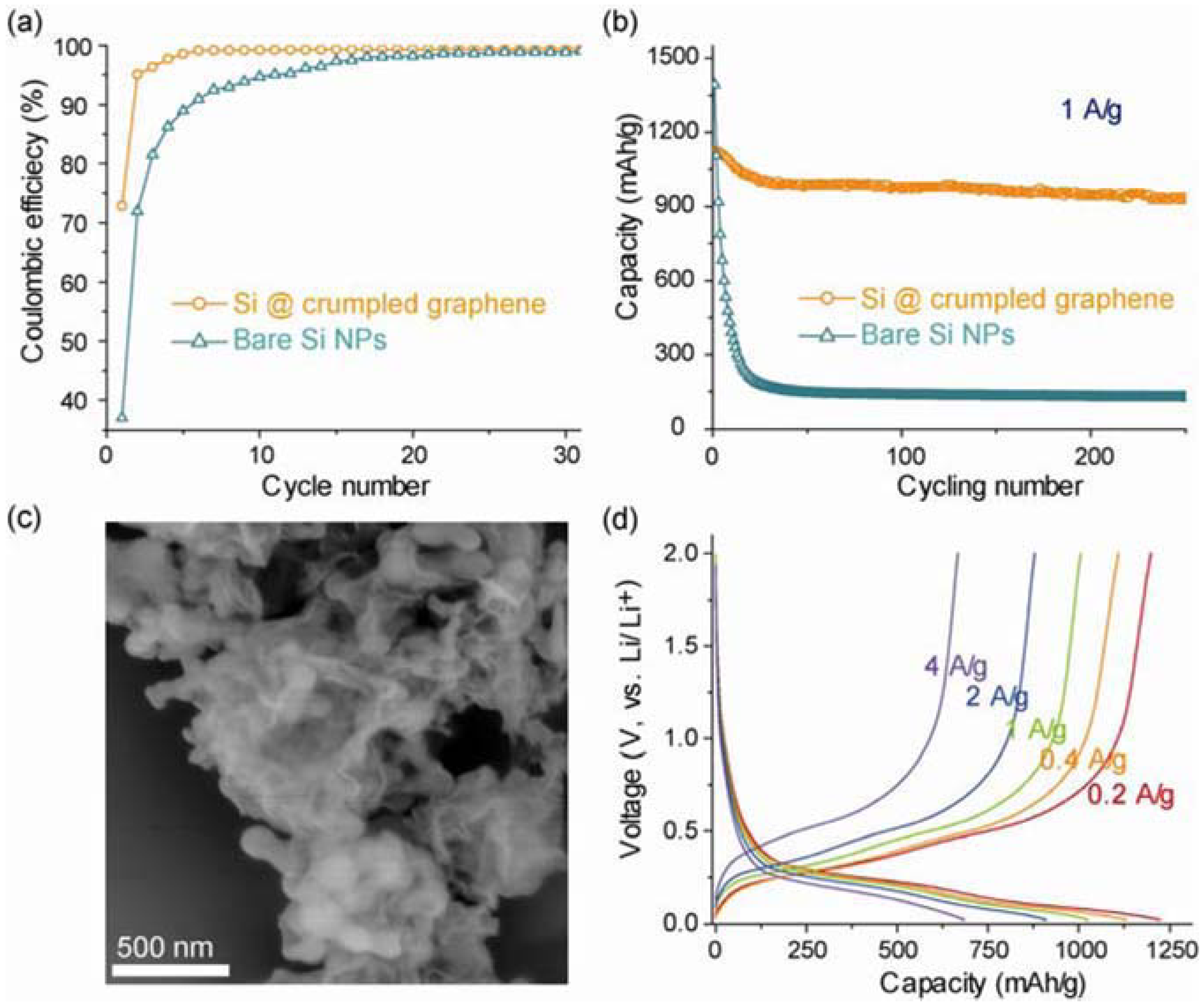
3.2. Silicon-Based Materials as Advanced Anode Materials for Lithium Ion Batteries
3.2.1. Pure Silicon Anode
3.2.2. Silicon/Carbon Composite Anode
3.2.3. New Binder Improved Silicon Performance
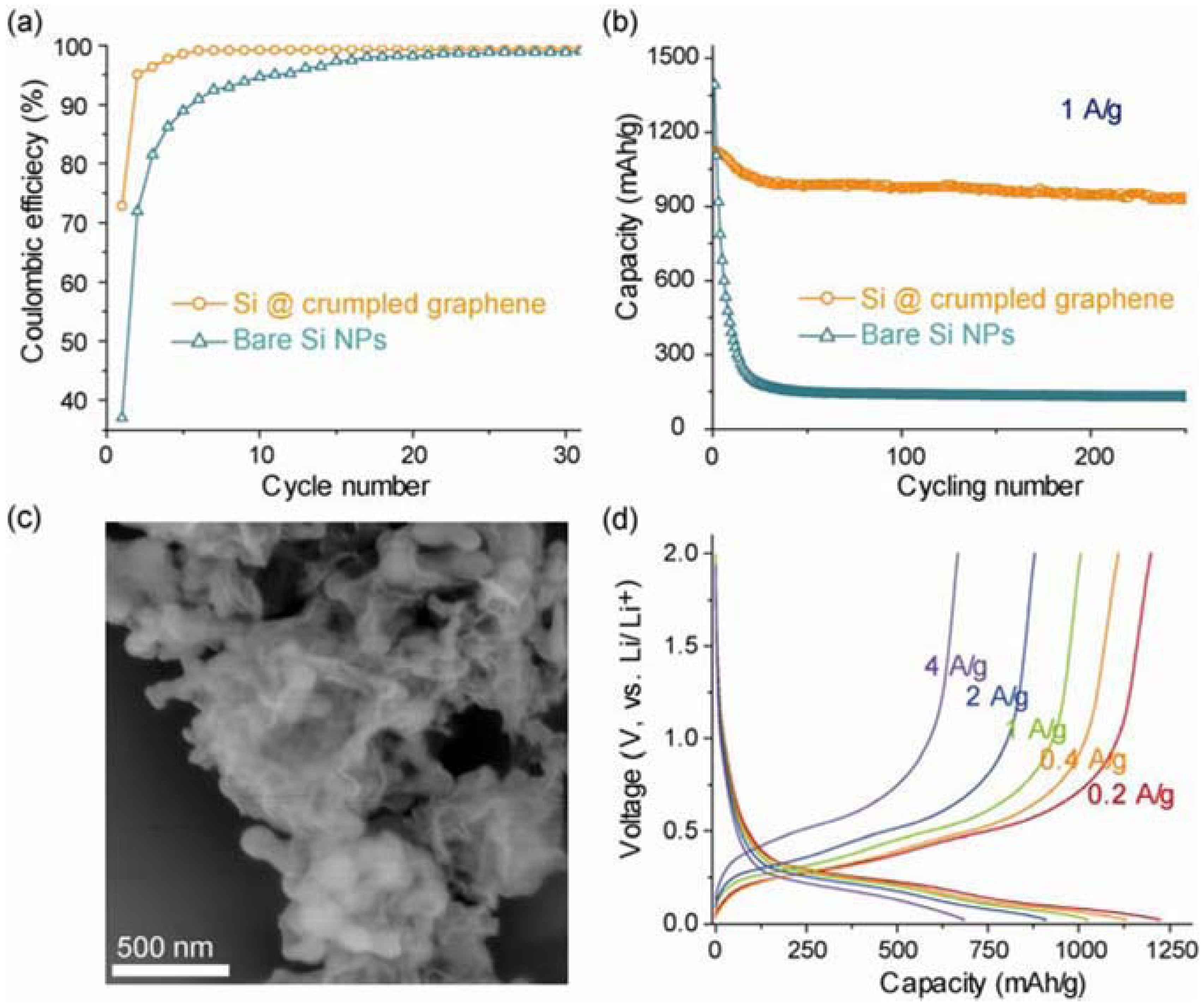
4. Current & Future Developments
Acknowledgments
References
- Whittingham, M.S. Electrical energy storage and intercalation chemistry. Science 1976, 192, 1126–1127. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, M.S. Lithium batteries and cathode materials. Chem. Rev. 2004, 104, 4271–4301. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, M.S. Materials challenges facing electrical energy storage. MRS Bull. 2008, 33, 411–419. [Google Scholar] [CrossRef]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. A review of nanostructured lithium ion battery materials via low temperature synthesis. Recent Pat. Nanotechnol. 2013, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Huang, H.; Yin, S.-C.; Nazar, L.F. Approaching theoretical capacity of LiFePO4 at room temperature at high rates. Electrochem. Solid-State Lett. 2001, 4, A170–A172. [Google Scholar] [CrossRef]
- Yang, S.; Song, Y.; Ngala, K.; Zavalij, P.Y.; Stanley Whittingham, M. Performance of LiFePO4 as lithium battery cathode and comparison with manganese and vanadium oxides. J. Power Sources 2003, 119–121, 239–246. [Google Scholar] [CrossRef]
- Chung, S.-Y.; Bloking, J.T.; Chiang, Y.-M. Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mater. 2002, 1, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Delacourt, C.; Poizot, P.; Levasseur, S.; Masquelier, C. Size effects on carbon-free LiFePO4 powders: The key to superior energy density. Electrochem. Solid-State Lett. 2006, 9, A352–A355. [Google Scholar] [CrossRef]
- Gibot, P.; Casas-Cabanas, M.; Laffont, L.; Levasseur, S.; Carlach, P.; Hamelet, S.; Tarascon, J.-M.; Masquelier, C. Room-temperature single-phase Li insertion/extraction in nanoscale LixFePO4. Nat. Mater. 2008, 7, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Armand, M.; Goodenough, J.B.; Padhi, A.K.; Nanjundaswamy, K.S.; Masquelier, C. Cathode Materials for Secondary (Rechargeable) Lithium Batteries. U.S. Patent 6,514,640, 4 February 2003. [Google Scholar]
- Chen, J.J.; Whittingham, M.S. Hydrothermal synthesis of lithium iron phosphate. Electrochem. Commun. 2006, 8, 855–858. [Google Scholar] [CrossRef]
- Chen, J.J.; Vacchio, M.J.; Wang, S.J.; Chernova, N.; Zavalij, P.Y.; Whittingham, M.S. The hydrothermal synthesis and characterization of olivines and related compounds for electrochemical applications. Solid State Ionics 2008, 178, 1676–1693. [Google Scholar] [CrossRef]
- Malik, R.; Burch, D.; Bazant, M.; Ceder, G. Particle size dependence of the ionic diffusivity. Nano Lett. 2010, 10, 4123–4127. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Graetz, J. Study of antisite defects in hydrothermally prepared LiFePO4 by in situ X-ray diffraction. ACS Appl. Mater. Interfaces 2011, 3, 1380–1384. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bai, J.; Chen, H.; Graetz, J. In situ hydrothermal synthesis of LiFePO4 studied by synchrotron X-ray diffraction. J. Phys. Chem. Lett. 2011, 2, 1874–1878. [Google Scholar] [CrossRef]
- Recham, N.; Oró-Solé, J.; Djellab, K.; Palacín, M.R.; Masquelier, C.; Tarascon, J.M. Hydrothermal synthesis, silver decoration and electrochemistry of LiMPO4 (M = Fe, Mn and Co) single crystals. Solid State Ionics 2012, 220, 47–52. [Google Scholar] [CrossRef]
- Doeff, M.M.; Chen, J.; Conry, T.E.; Wang, R.; Wilcox, J.; Aumentado, A. Combustion synthesis of nanoparticulate LiMgxMn1 − xPO4 (x = 0, 0.1, 0.2) carbon composites. J. Mater. Res. 2010, 25, 1460–1468. [Google Scholar] [CrossRef]
- Sun, Y.-K.; Oh, S.-M.; Park, H.-K.; Scrosati, B. Micrometer-sized, nanoporous, high-volumetric-capacity LiMn0.85Fe0.15PO4 cathode material for rechargeable lithium-ion batteries. Adv. Mater. 2011, 23, 5050–5054. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-M.; Oh, S.-W.; Yoon, C.-S.; Scrosati, B.; Amine, K.; Sun, Y.-K. High-performance carbon-LiMnPO4 nanocomposite cathode for lithium batteries. Adv. Funct. Mater. 2010, 20, 3260–3265. [Google Scholar] [CrossRef]
- Yoo, H.; Jo, M.; Jin, B.-S.; Kim, H.-S.; Cho, J. Flexible morphology design of 3D-macroporous LiMnPO4 cathode materials for Li secondary batteries: Ball to flake. Adv. Energy Mater. 2011, 1, 347–351. [Google Scholar] [CrossRef]
- Ni, J.; Wang, H.; Gao, L.; Lu, L. A high-performance LiCoPO4/C core/shell composite for Li-ion batteries. Electrochim. Acta 2012, 70, 349–354. [Google Scholar] [CrossRef]
- Oh, S.-M.; Myung, S.-T.; Sun, Y.-K. Olivine LiCoPO4-carbon composite showing high rechargeable capacity. J. Mater. Chem. 2012, 22, 14932–14937. [Google Scholar] [CrossRef]
- Sharabi, R.; Markevich, E.; Borgel, V.; Salitra, G.; Aurbach, D.; Semrau, G.; Schmidt, M.A.; Schall, N.; Stinner, C. Significantly improved cycling performance of LiCoPO4 cathodes. Electrochem. Commun. 2011, 13, 800–802. [Google Scholar] [CrossRef]
- Dominko, R. Li2MSiO4 (M = Fe and/or Mn) cathode materials. J. Power Sources 2008, 184, 462–468. [Google Scholar] [CrossRef]
- Nytén, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J.O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 2005, 7, 156–160. [Google Scholar] [CrossRef]
- Nyten, A.; Kamali, S.; Haggstrom, L.; Gustafsson, T.; Thomas, J.O. The lithium extraction/insertion mechanism in Li2FeSiO4. J. Mater. Chem. 2006, 16, 2266–2272. [Google Scholar] [CrossRef]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and lithium transport pathways in Li2FeSiO4 cathodes for lithium batteries. J. Am. Chem. Soc. 2011, 133, 13031–13035. [Google Scholar] [CrossRef] [PubMed]
- Liivat, A.; Thomas, J.O. Li-ion migration in Li2FeSiO4-related cathode materials: A DFT study. Solid State Ionics 2011, 192, 58–64. [Google Scholar] [CrossRef]
- Belharouak, I.; Abouimrane, A.; Amine, K. Structural and electrochemical characterization of Li2MnSiO4 cathode material. J. Phys. Chem. C 2009, 113, 20733–20737. [Google Scholar] [CrossRef]
- Gong, Z.L.; Li, Y.X.; He, G.N.; Li, J.; Yang, Y. Nanostructured Li2FeSiO4 electrode material synthesized through hydrothermal-assisted sol-gel process. Electrochem. Solid-State Lett. 2008, 11, A60–A63. [Google Scholar] [CrossRef]
- Kokalj, A.; Dominko, R.; Mali, G.; Meden, A.; Gaberscek, M.; Jamnik, J. Beyond one-electron reaction in Li cathode materials: Designing Li2MnxFe1 − xSiO4. Chem. Mater. 2007, 19, 3633–3640. [Google Scholar] [CrossRef]
- Aravindan, V.; Karthikeyan, K.; Ravi, S.; Amaresh, S.; Kim, W.S.; Lee, Y.S. Adipic acid assisted sol-gel synthesis of Li2MnSiO4 nanoparticles with improved lithium storage properties. J. Mater. Chem. 2010, 20, 7340–7343. [Google Scholar] [CrossRef]
- Rangappa, D.; Murukanahally, K.D.; Tomai, T.; Unemoto, A.; Honma, I. Ultrathin nanosheets of Li2MSiO4 (M = Fe, Mn) as high-capacity Li-Ion battery electrode. Nano Lett. 2012, 12, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-de Dompablo, M.E.; Armand, M.; Tarascon, J.M.; Amador, U. On-demand design of polyoxianionic cathode materials based on electronegativity correlations: An exploration of the Li2MSiO4 system (M = Fe, Mn, Co, Ni). Electrochem. Commun. 2006, 8, 1292–1298. [Google Scholar]
- Gong, Z.L.; Li, Y.X.; Yang, Y. Synthesis and electrochemical performance of Li2CoSiO4 as cathode material for lithium ion batteries. J. Power Sources 2007, 174, 524–527. [Google Scholar] [CrossRef]
- Lyness, C.; Delobel, B.; Armstrong, A.R.; Bruce, P.G. The lithium intercalation compound Li2CoSiO4 and its behaviour as a positive electrode for lithium batteries. Chem. Commun. 2007, 4890–4892. [Google Scholar]
- Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.M. Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries. Nature 2000, 407, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.M. Searching for new anode materials for the Li-ion technology: time to deviate from the usual path. J. Power Sources 2001, 97–98, 235–239. [Google Scholar] [CrossRef]
- Bruce, P.G.; Scrosati, B.; Tarascon, J.-M. Nanomaterials for rechargeable lithium batteries. Angew. Chem. Int. Ed. 2008, 47, 2930–2946. [Google Scholar] [CrossRef]
- Wu, H.B.; Chen, J.S.; Hng, H.H.; Wen Lou, X. Nanostructured metal oxide-based materials as advanced anodes for lithium-ion batteries. Nanoscale 2012, 4, 2526–2542. [Google Scholar] [CrossRef] [PubMed]
- Goward, G.R.; Taylor, N.J.; Souza, D.C.S.; Nazar, L.F. The true crystal structure of Li17M4 (M = Ge, Sn, Pb)–revised from Li22M5. J. Alloys Compd. 2001, 329, 82–91. [Google Scholar] [CrossRef]
- Lupu, C.; Mao, J.-G.; Rabalais, J.W.; Guloy, A.M.; Richardson, J.W. X-ray and neutron diffraction studies on “Li4.4Sn”. Inorg. Chem. 2003, 42, 3765–3771. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zavalij, P.Y.; Whittingham, M.S. Anodes for lithium batteries: Tin revisited. Electrochem. Commun. 2003, 5, 587–590. [Google Scholar] [CrossRef]
- Egashira, M.; Takatsuji, H.; Okada, S.; Yamaki, J.-i. Properties of containing Sn nanoparticles activated carbon fiber for a negative electrode in lithium batteries. J. Power Sources 2002, 107, 56–60. [Google Scholar] [CrossRef]
- Morishita, T.; Hirabayashi, T.; Okuni, T.; Ota, N.; Inagaki, M. Preparation of carbon-coated Sn powders and their loading onto graphite flakes for lithium ion secondary battery. J. Power Sources 2006, 160, 638–644. [Google Scholar] [CrossRef]
- Kim, I.-s.; Blomgren, G.E.; Kumta, P.N. Sn/C composite anodes for Li-ion batteries. Electrochem. Solid-State Lett. 2004, 7, A44–A48. [Google Scholar] [CrossRef]
- Hassoun, J.; Panero, S.; Simon, P.; Taberna, P.L.; Scrosati, B. High-rate, long-life Ni–Sn nanostructured electrodes for lithium-ion batteries. Adv. Mater. 2007, 19, 1632–1635. [Google Scholar] [CrossRef]
- Luo, B.; Wang, B.; Liang, M.; Ning, J.; Li, X.; Zhi, L. Reduced graphene oxide-mediated growth of uniform tin-core/carbon-sheath coaxial nanocables with enhanced lithium ion storage properties. Adv. Mater. 2012, 24, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Chupas, P.J.; Whittingham, M.S. Characterization of amorphous and crystalline tin–cobalt anodes. Electrochem. Solid-State Lett. 2007, 10, A274–A278. [Google Scholar] [CrossRef]
- Zhang, R.; Whittingham, M.S. Electrochemical behavior of the amorphous tin–cobalt anode. Electrochem. Solid-State Lett. 2010, 13, A184–A187. [Google Scholar] [CrossRef]
- Idota, Y.; Kubota, T.; Matsufuji, A.; Maekawa, Y.; Miyasaka, T. Tin-based amorphous oxide: A high-capacity lithium-ion-storage material. Science 1997, 276, 1395–1397. [Google Scholar] [CrossRef]
- Kim, C.; Noh, M.; Choi, M.; Cho, J.; Park, B. Critical size of a nano SnO2 electrode for Li-secondary battery. Chem. Mater. 2005, 17, 3297–3301. [Google Scholar] [CrossRef]
- Wang, X.-L.; Feygenson, M.; Aronson, M.C.; Han, W.-Q. Sn/SnOx core−shell nanospheres: Synthesis, anode performance in li-ion batteries and superconductivity. J. Phys. Chem. C 2010, 114, 14697–14703. [Google Scholar] [CrossRef]
- Lou, X.W.; Wang, Y.; Yuan, C.; Lee, J.Y.; Archer, L.A. Template-free synthesis of SnO2 hollow nanostructures with high lithium storage capacity. Adv. Mater. 2006, 18, 2325–2329. [Google Scholar] [CrossRef]
- Deng, D.; Lee, J.Y. Hollow core–shell mesospheres of crystalline SnO2 nanoparticle aggregates for high capacity Li+ ion storage. Chem. Mater. 2008, 20, 1841–1846. [Google Scholar] [CrossRef]
- Wang, Z.; Luan, D.; Boey, F.Y.C.; Lou, X.W. Fast formation of SnO2 nanoboxes with enhanced lithium storage capability. J. Am. Chem. Soc. 2011, 133, 4738–4741. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lee, J.Y.; Zeng, H.C. Polycrystalline SnO2 nanotubes prepared via infiltration casting of nanocrystallites and their electrochemical application. Chem. Mater. 2005, 17, 3899–3903. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, H.; Yang, R.; Li, X.; Qi, L. Morphology-controlled synthesis of SnO2 nanotubes by using 1D silica mesostructures as sacrificial templates and their applications in lithium-ion batteries. Small 2010, 6, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-S.; Wang, G.-X.; Kang, Y.-M.; Wexler, D.; Dou, S.-X.; Liu, H.-K. Preparation and electrochemical properties of SnO2 nanowires for application in lithium-ion batteries. Angew. Chem. Int. Ed. 2007, 46, 750–753. [Google Scholar] [CrossRef]
- Park, M.-S.; Kang, Y.-M.; Wang, G.-X.; Dou, S.-X.; Liu, H.-K. The effect of morphological modification on the electrochemical properties of SnO2 nanomaterials. Adv. Funct. Mater. 2008, 18, 455–461. [Google Scholar] [CrossRef]
- Wu, H.B.; Chen, J.S.; Lou, X.W.; Hng, H.H. Synthesis of SnO2 hierarchical structures assembled from nanosheets and their lithium storage properties. J. Phys. Chem. C 2011, 115, 24605–24610. [Google Scholar] [CrossRef]
- Chen, J.S.; Ng, M.F.; Wu, H.B.; Zhang, L.; Lou, X.W. Synthesis of phase-pure SnO2 nanosheets with different organized structures and their lithium storage properties. CrystEngComm 2012, 14, 5133–5136. [Google Scholar] [CrossRef]
- Chen, J.; Wang, S.; Whittingham, M.S. Hydrothermal synthesis of cathode materials. J. Power Sources 2007, 174, 442–448. [Google Scholar] [CrossRef]
- Lee, K.T.; Jung, Y.S.; Oh, S.M. Synthesis of tin-encapsulated spherical hollow carbon for anode material in lithium secondary batteries. J. Am. Chem. Soc. 2003, 125, 5652–5653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-M.; Hu, J.-S.; Guo, Y.-G.; Zheng, S.-F.; Zhong, L.-S.; Song, W.-G.; Wan, L.-J. Tin-nanoparticles encapsulated in elastic hollow carbon spheres for high-performance anode material in lithium-ion batteries. Adv. Mater. 2008, 20, 1160–1165. [Google Scholar] [CrossRef]
- Lou, X.W.; Chen, J.S.; Chen, P.; Archer, L.A. One-pot synthesis of carbon-coated SnO2 nanocolloids with improved reversible lithium storage properties. Chem. Mater. 2009, 21, 2868–2874. [Google Scholar] [CrossRef]
- Wang, D.; Kou, R.; Choi, D.; Yang, Z.; Nie, Z.; Li, J.; Saraf, L.V.; Hu, D.; Zhang, J.; Graff, G.L.; Liu, J.; Pope, M.A.; Aksay, I.A. Ternary self-assembly of ordered metal oxide-graphene nanocomposites for electrochemical energy storage. ACS Nano 2010, 4, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-S.; Jiang, L.-Y.; Yan, H.-J.; Wang, W.D.; Wang, W.; Song, W.-G.; Guo, Y.-G.; Wan, L.-J. Mono dispersed SnO2 nanoparticles on both sides of single layer graphene sheets as anode materials in Li-ion batteries. J. Mater. Chem. 2010, 20, 5462–5467. [Google Scholar] [CrossRef]
- Ding, S.; Luan, D.; Boey, F.Y.C.; Chen, J.S.; Lou, X.W. SnO2 nanosheets grown on graphene sheets with enhanced lithium storage properties. Chem. Commun. 2011, 47, 7155–7157. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, H.C.; Lee, J.Y. Highly reversible lithium storage in porous SnO2 nanotubes with coaxially grown carbon nanotube overlayers. Adv. Mater. 2006, 18, 645–649. [Google Scholar] [CrossRef]
- Wen, Z.; Wang, Q.; Zhang, Q.; Li, J. In situ growth of mesoporous SnO2 on multiwalled carbon nanotubes: A novel composite with porous-tube structure as anode for lithium batteries. Adv. Funct. Mater. 2007, 17, 2772–2778. [Google Scholar] [CrossRef]
- Huang, J.Y.; Zhong, L.; Wang, C.M.; Sullivan, J.P.; Xu, W.; Zhang, L.Q.; Mao, S.X.; Hudak, N.S.; Liu, X.H.; Subramanian, A.; Fan, H.; Qi, L.; Kushima, A.; Li, J. In situ observation of the electrochemical lithiation of a single SnO2 nanowire electrode. Science 2010, 330, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Han, W.-Q.; Chen, J.; Graetz, J. Single-crystal intermetallic M−Sn (M = Fe, Cu, Co, Ni) nanospheres as negative electrodes for lithium-ion batteries. ACS Appl. Mater. Interfaces 2010, 2, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Feygenson, M.; Chen, H.; Lin, C.-H.; Ku, W.; Bai, J.; Aronson, M.C.; Tyson, T.A.; Han, W.-Q. Nanospheres of a new intermetallic FeSn5 phase: synthesis, magnetic properties and anode performance in li-ion batteries. J. Am. Chem. Soc. 2011, 133, 11213–11219. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, B.A.; Lesh, G.C.; Huggins, R.A. All-solid lithium electrodes with mixed-conductor matrix. J. Electrochem. Soc. 1981, 128, 725–729. [Google Scholar] [CrossRef]
- Kasavajjula, U.; Wang, C.; Appleby, A.J. Nano- and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J. Power Sources 2007, 163, 1003–1039. [Google Scholar] [CrossRef]
- Zhang, W.-J. A review of the electrochemical performance of alloy anodes for lithium-ion batteries. J. Power Sources 2011, 196, 13–24. [Google Scholar] [CrossRef]
- Ryu, J.H.; Kim, J.W.; Sung, Y.-E.; Oh, S.M. Failure modes of silicon powder negative electrode in lithium secondary batteries. Electrochem. Solid-State Lett. 2004, 7, A306–A309. [Google Scholar] [CrossRef]
- Li, H.; Huang, X.; Chen, L.; Wu, Z.; Liang, Y. A High capacity nano Si composite anode material for lithium rechargeable batteries. Electrochem. Solid-State Lett. 1999, 2, 547–549. [Google Scholar] [CrossRef]
- Kim, H.; Seo, M.; Park, M.-H.; Cho, J. A Critical size of silicon nano-anodes for lithium rechargeable batteries. Angew. Chem. Int. Ed. 2010, 49, 2146–2149. [Google Scholar] [CrossRef]
- Wang, C.S.; Wu, G.T.; Zhang, X.B.; Qi, Z.F.; Li, W.Z. Lithium insertion in carbon-silicon composite materials produced by mechanical milling. J. Electrochem. Soc. 1998, 145, 2751–2758. [Google Scholar] [CrossRef]
- Magasinski, A.; Dixon, P.; Hertzberg, B.; Kvit, A.; Ayala, J.; Yushin, G. High-performance lithium-ion anodes using a hierarchical bottom-up approach. Nat Mater 2010, 9, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Smith, K.B.; Hayner, C.M.; Kung, H.H. Silicon nanoparticles-graphene paper composites for Li-ion battery anodes. Chem. Commun. 2010, 46, 2025–2027. [Google Scholar] [CrossRef]
- Luo, J.; Zhao, X.; Wu, J.; Jang, H.D.; Kung, H.H.; Huang, J. Crumpled graphene-encapsulated Si nanoparticles for lithium ion battery anodes. J. Phys. Chem. Lett. 2012, 3, 1824–1829. [Google Scholar] [CrossRef]
- Kovalenko, I.; Zdyrko, B.; Magasinski, A.; Hertzberg, B.; Milicev, Z.; Burtovyy, R.; Luzinov, I.; Yushin, G. A Major constituent of brown algae for use in high-capacity Li-ion batteries. Science 2011, 334, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, J.; Bucur, C.B.; Oliver, A.G.; Sugimoto, T.; Matsui, M.; Kim, H.S.; Allred, G.D.; Zajicek, J.; Kotani, Y. Electrolyte roadblocks to a magnesium rechargeable battery. Energy Environ. Sci. 2012, 5, 5941–5950. [Google Scholar] [CrossRef]
- Kim, H.S.; Arthur, T.S.; Allred, G.D.; Zajicek, J.; Newman, J.G.; Rodnyansky, A.E.; Oliver, A.G.; Boggess, W.C.; Muldoon, J. Structure and compatibility of a magnesium electrolyte with a sulphur cathode. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Mohtadi, R.; Matsui, M.; Arthur, T.S.; Hwang, S.-J. Magnesium borohydride: From hydrogen storage to magnesium battery. Angew. Chem. Int. Ed. 2012, 51, 9780–9783. [Google Scholar] [CrossRef]
- Zhang, R.; Yu, X.; Nam, K.-W.; Ling, C.; Arthur, T.S.; Song, W.; Knapp, A.M.; Ehrlich, S.N.; Yang, X.-Q.; Matsui, M. α-MnO2 as a cathode material for rechargeable Mg batteries. Electrochem. Commun. 2012, 23, 110–113. [Google Scholar] [CrossRef]
- Arthur, T.S.; Singh, N.; Matsui, M. Electrodeposited Bi, Sb and Bi1 − xSbx alloys as anodes for Mg-ion batteries. Electrochem. Commun. 2012, 16, 103–106. [Google Scholar] [CrossRef]
- Singh, N.; Arthur, T.S.; Ling, C.; Matsui, M.; Mizuno, F. A high energy-density tin anode for rechargeable magnesium-ion batteries. Chem. Commun. 2013, 49, 149–151. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, J. Recent Progress in Advanced Materials for Lithium Ion Batteries. Materials 2013, 6, 156-183. https://doi.org/10.3390/ma6010156
Chen J. Recent Progress in Advanced Materials for Lithium Ion Batteries. Materials. 2013; 6(1):156-183. https://doi.org/10.3390/ma6010156
Chicago/Turabian StyleChen, Jiajun. 2013. "Recent Progress in Advanced Materials for Lithium Ion Batteries" Materials 6, no. 1: 156-183. https://doi.org/10.3390/ma6010156
APA StyleChen, J. (2013). Recent Progress in Advanced Materials for Lithium Ion Batteries. Materials, 6(1), 156-183. https://doi.org/10.3390/ma6010156