The Effect of PtRuIr Nanoparticle Crystallinity in Electrocatalytic Methanol Oxidation

 ,
,
Abstract
:1. Introduction
2. Results and Discussion

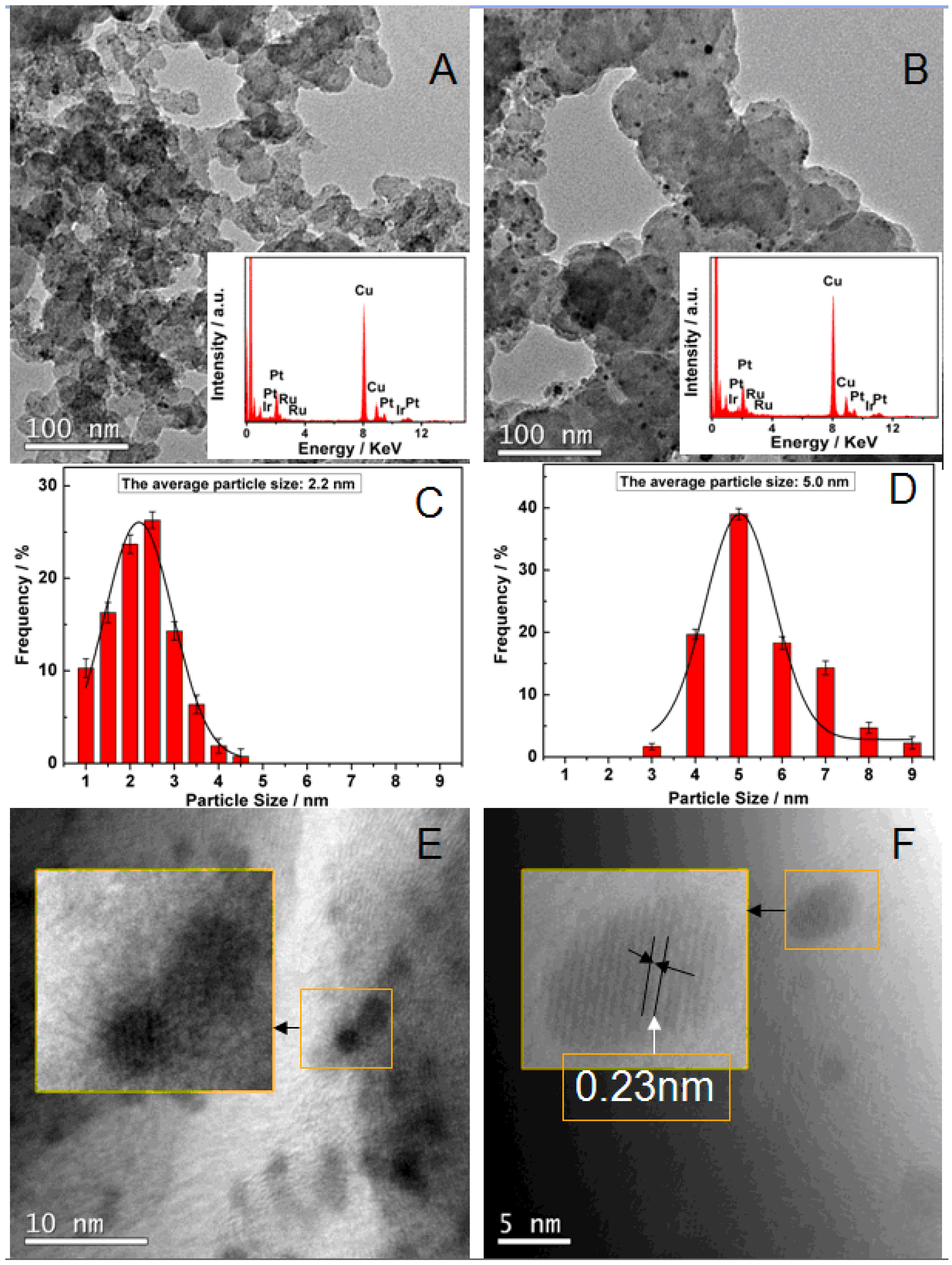

{kind=link}
{kind=link}
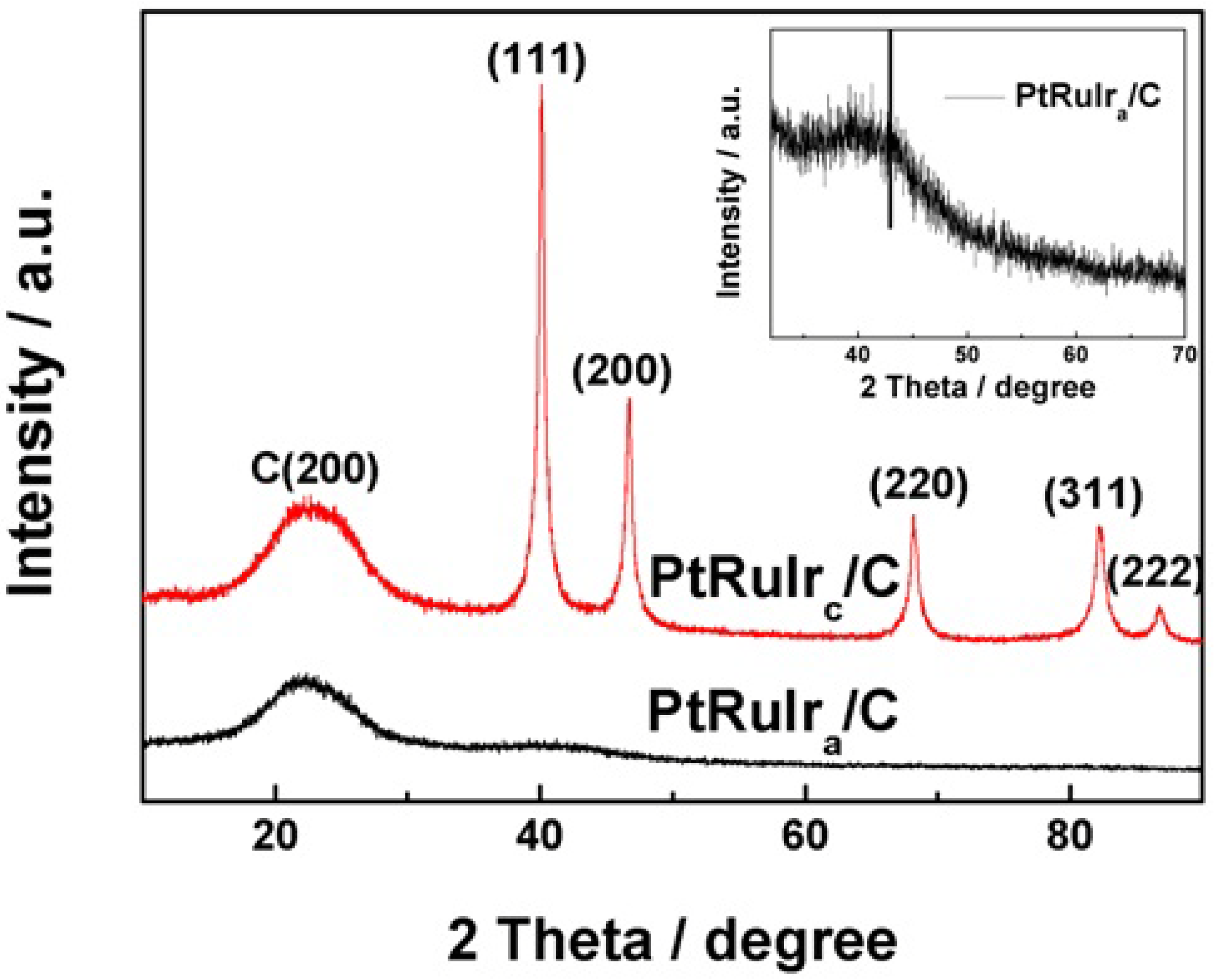
{kind=link}
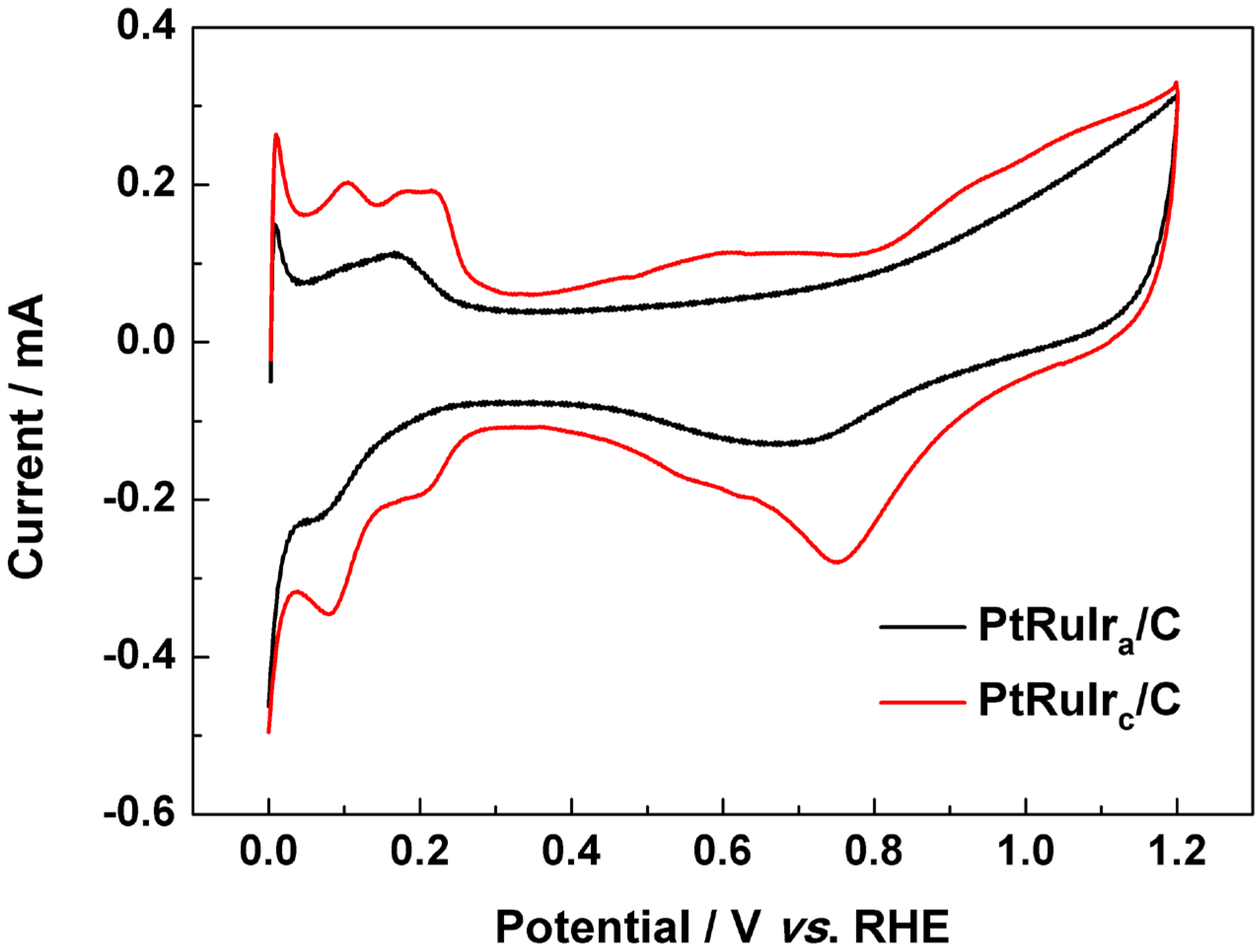
{kind=link}
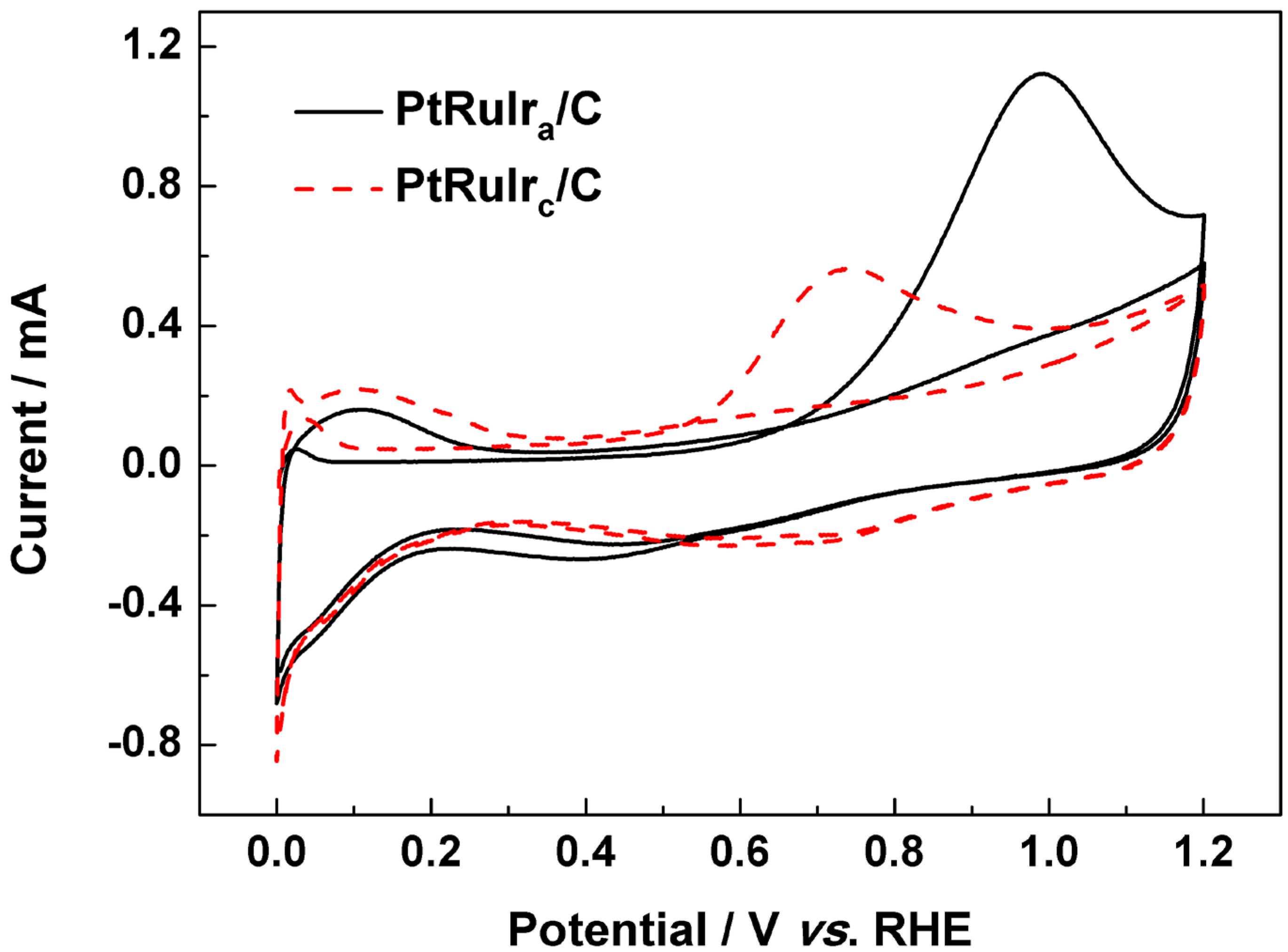
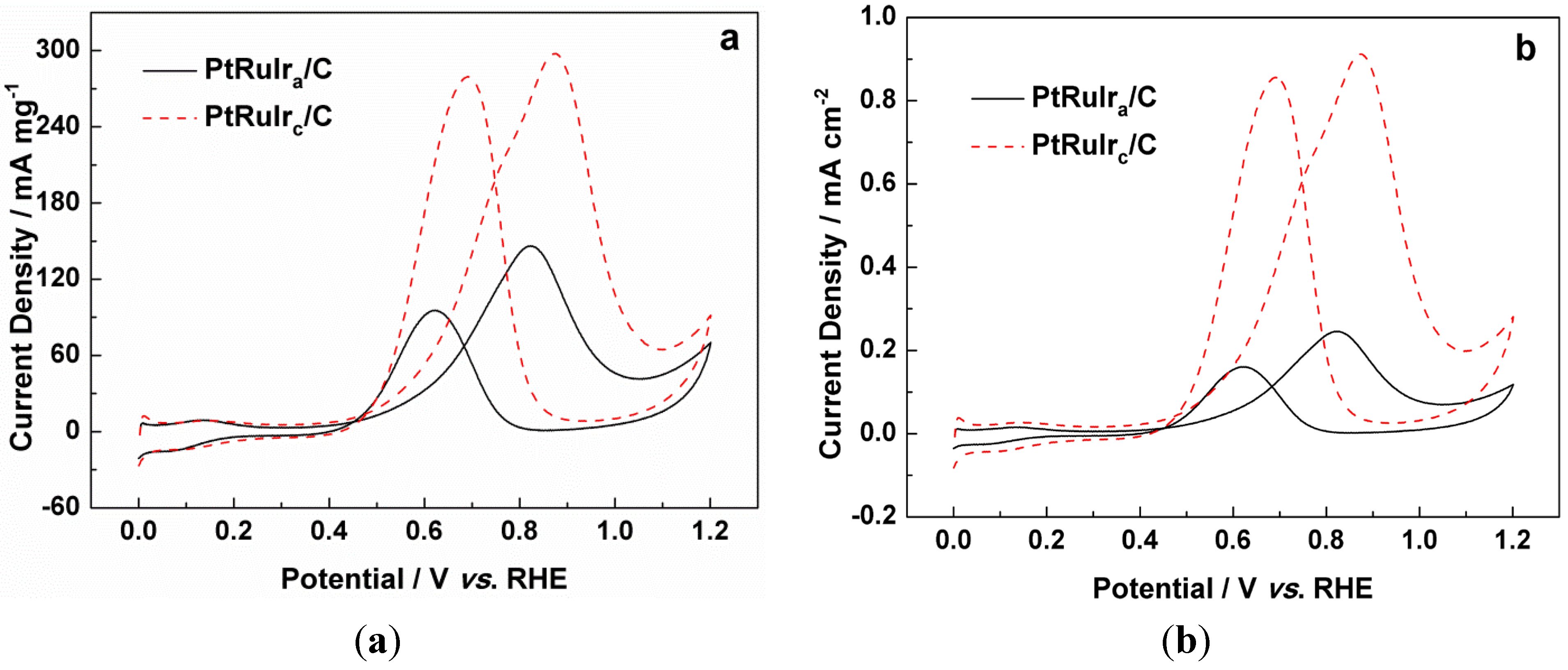{kind=link}
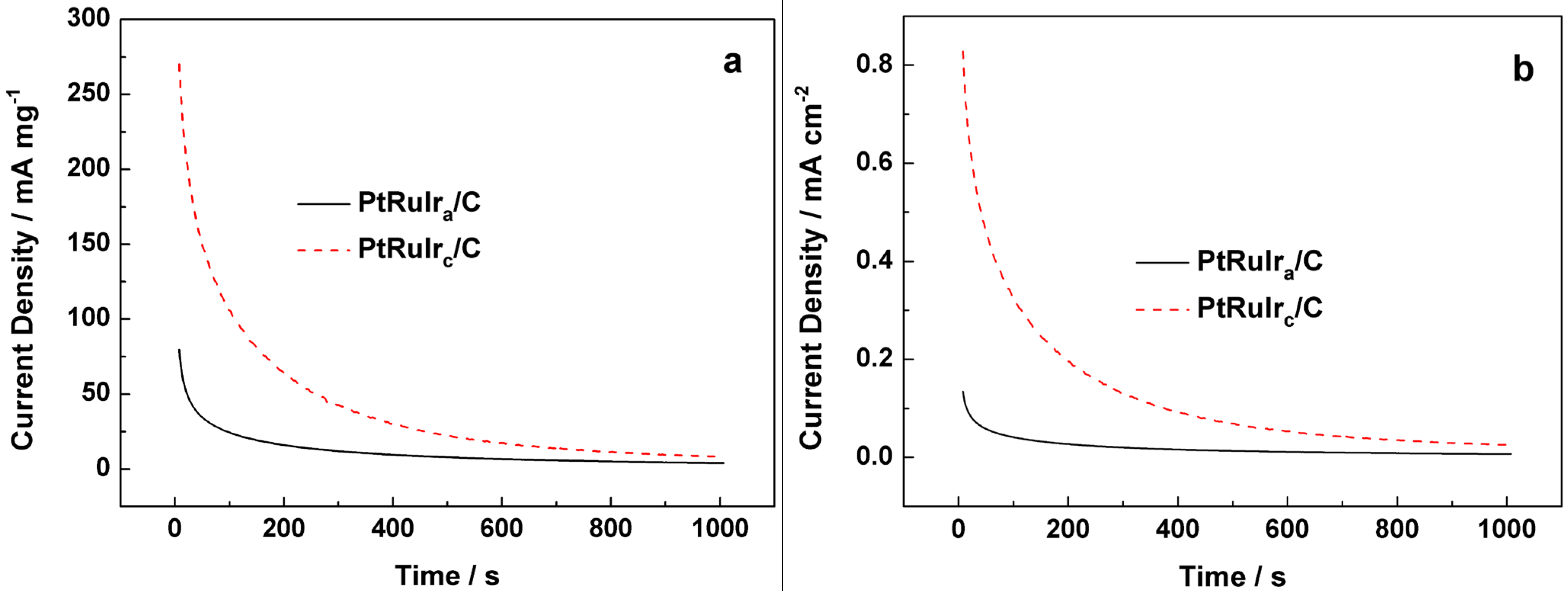{kind=link}
| Catalyst | PtRuIra/C | PtRuIrc/C |
|---|---|---|
| Pt:Ru:Ir atom ratio | 3.5:3.0:1.0 | 3.5:3.0:1.0 |
| The average particle size/nm | 2.2 ± 0.02 | 5.0 ± 0.02 |
| ECSA/m2 g−1metal | 59.5 | 32.6 |
| The onset potential for CO oxidation/mV vs. RHE | 663 | 521 |
| The onset potential for methanol oxidation/mV vs. RHE | 370 | 338 |
| The mass activity for methanol oxidation/mA mg−1 | 147 | 298 |
| The specific activity for methanol oxidation/mA cm−2 | 0.25 | 0.91 |
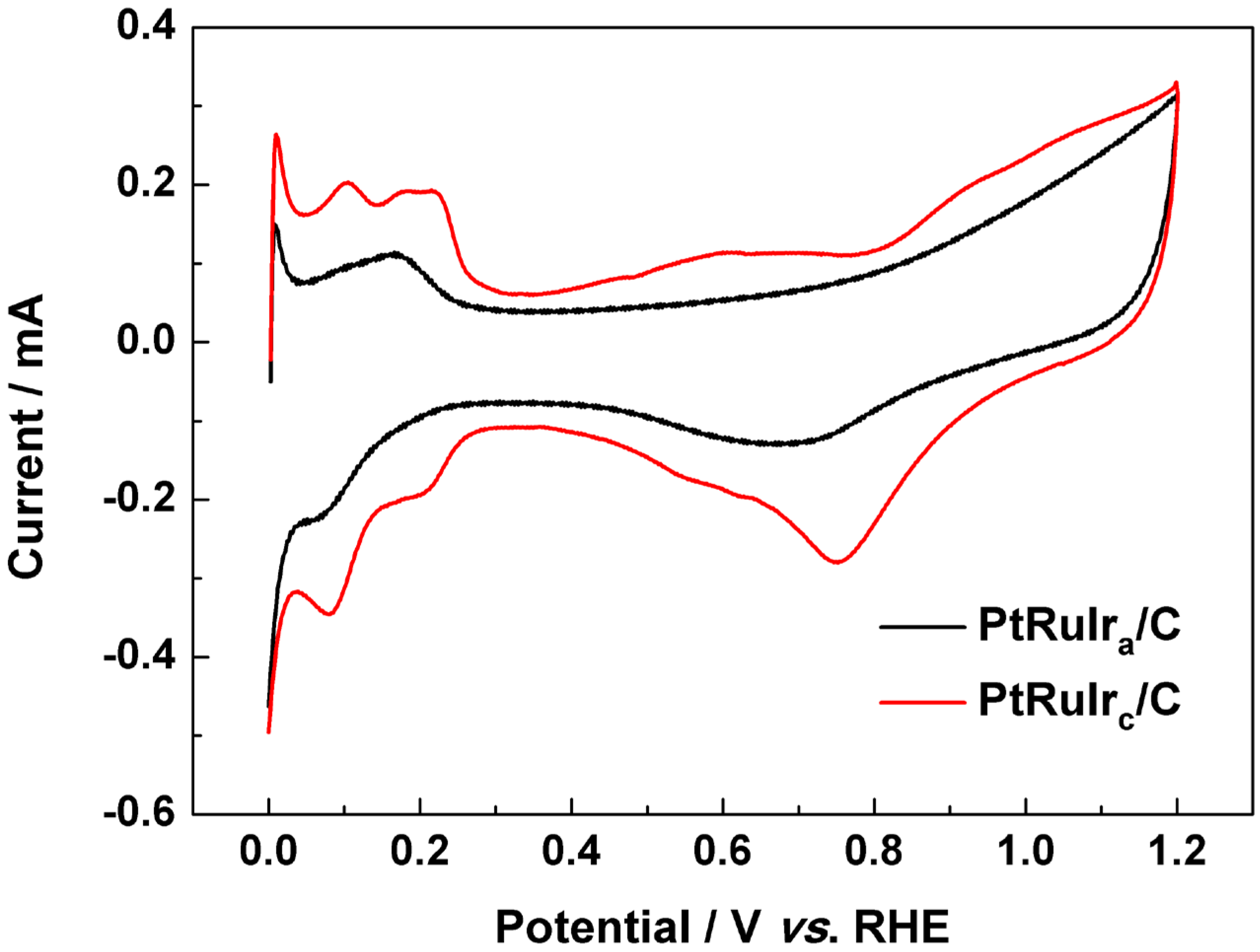
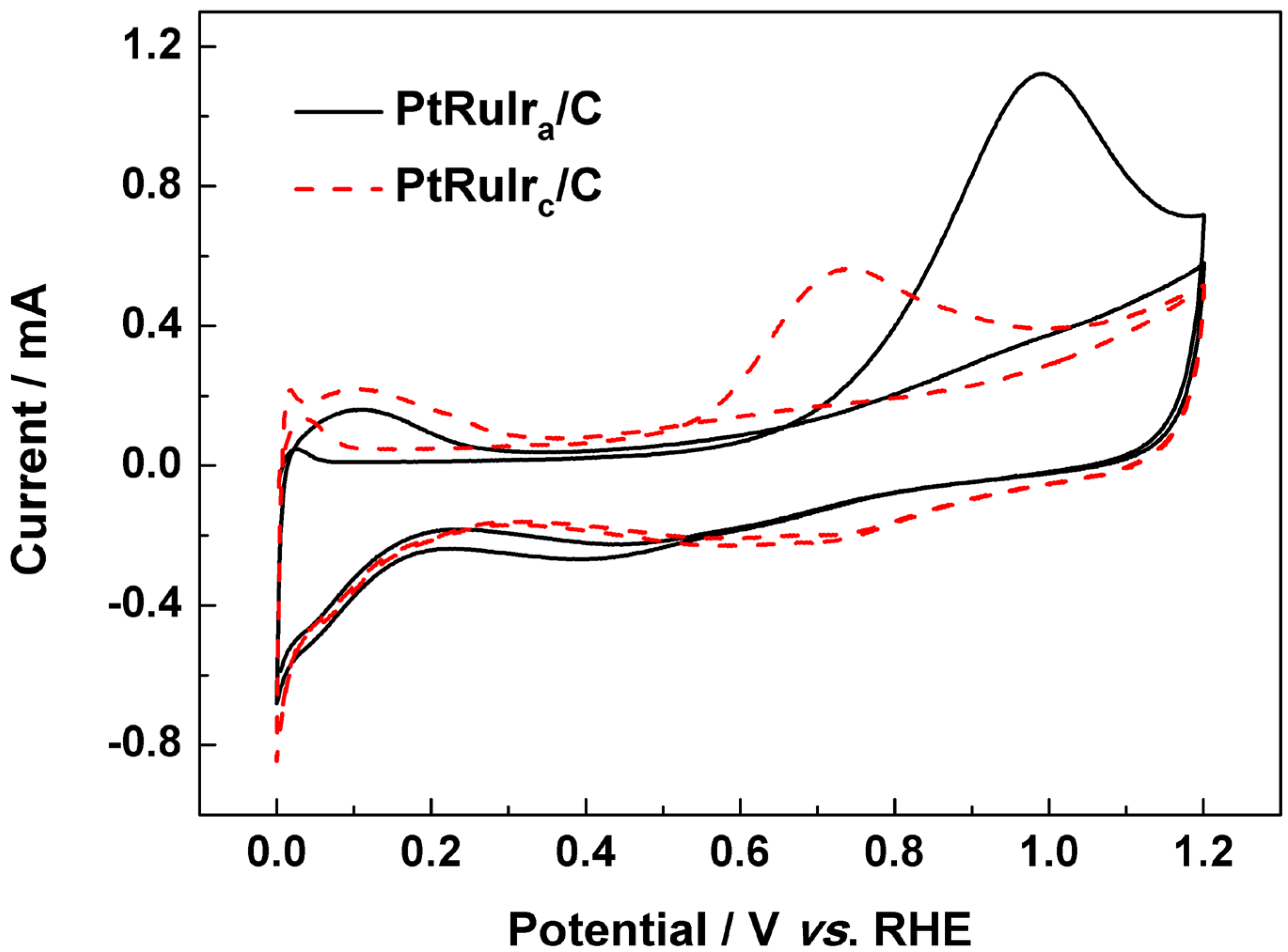
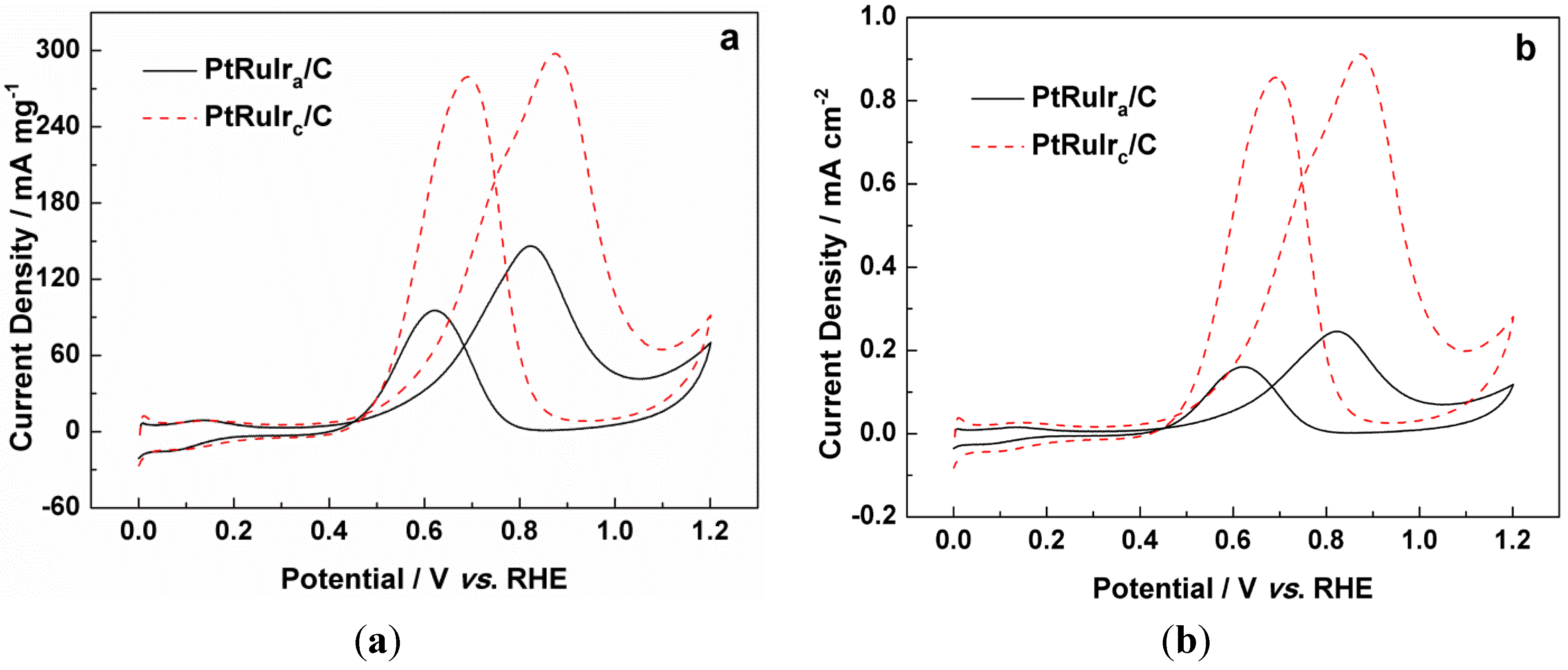
3. Experimental Section
3.1. Preparation of PtRuIr/C Catalysts with Different Crystallinity
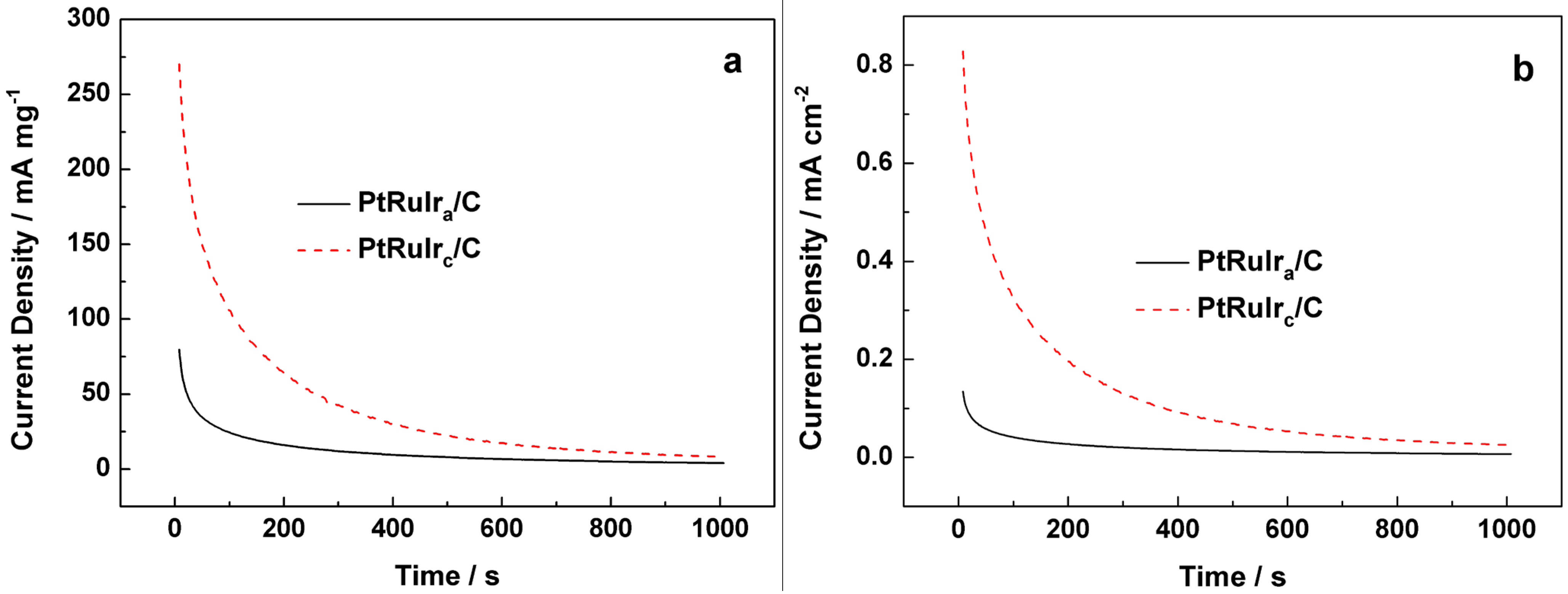
3.2. Measurements
4. Conclusions
Acknowledgments
References
- Peng, Z.; Yang, H. Designer platinum nanoparticles: Control of shape, composition in alloy, nanostructure and electrocatalytic property. Nano Today 2009, 4, 143–164. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, M.; Xia, Y. Noble-Metal Nanocrystals with Concave Surfaces: Synthesis and Applications. Angew. Chem. Int. Ed. 2012, 51, 7656–7673. [Google Scholar] [CrossRef]
- Rabis, A.; Rodriguez, P.; Schmidt, T.J. Electrocatalysis for Polymer Electrolyte Fuel Cells: Recent Achievements and Future Challenges. ACS Catal. 2012, 2, 864–890. [Google Scholar] [CrossRef]
- An, X.-S.; Fan, Y.-J.; Chen, D.-J.; Wang, Q.; Zhou, Z.-Y.; Sun, S.-G. Enhanced activity of rare earth doped PtRu/C catalysts for methanol electro-oxidation. Electrochim. Acta 2011, 56, 8912–8918. [Google Scholar] [CrossRef]
- Daimon, H.; Kurobe, Y. Size reduction of PtRu catalyst particle deposited on carbon support by addition of non-metallic elements. Catal. Today 2006, 111, 182–187. [Google Scholar] [CrossRef]
- Gómez de la Fuente, J.L.; Martínez-Huerta, M.V.; Rojas, S.; Hernández-Fernández, P.; Terreros, P.; Fierro, J.L.G.; Peña, M.A. Tailoring and structure of PtRu nanoparticles supported on functionalized carbon for DMFC applications: New evidence of the hydrous ruthenium oxide phase. Appl. Catal. B 2009, 88, 505–514. [Google Scholar] [CrossRef]
- Huang, T.; Wang, X.Y.; Zhuang, J.H.; Cai, W.B.; Yu, A.S. Preparation of Porous PtRuCo Catalyst by One-Step Codeposition and Its Electrocatalytic Performance for Methanol Oxidation. Electrochem. Solid State Lett. 2009, 12, B112–B115. [Google Scholar] [CrossRef]
- Zhu, J.; Cheng, F.; Tao, Z.; Chen, J. Electrocatalytic Methanol Oxidation of Pt0.5Ru0.5−xSnx/C (x = 0−0.5). J. Phys. Chem. C 2008, 112, 6337–6345. [Google Scholar] [CrossRef]
- Liao, S.; Holmes, K.-A.; Tsaprailis, H.; Birss, V.I. High Performance PtRuIr Catalysts Supported on Carbon Nanotubes for the Anodic Oxidation of Methanol. J. Am. Chem. Soc. 2006, 128, 3504–3505. [Google Scholar] [CrossRef] [PubMed]
- Khorasani-Motlagh, M.; Noroozifar, M.; Ekrami-Kakhki, M.-S. Investigation of the nanometals (Ni and Sn) in platinum binary and ternary electrocatalysts for methanol electrooxidation. Int. J. Hydrog. Energy 2011, 36, 11554–11563. [Google Scholar] [CrossRef]
- Eguiluz, K.I. B.; Salazar-Banda, G.R.; Miwa, D.; Machado, S.A. S.; Avaca, L.A. Effect of the catalyst composition in the Ptx(Ru–Ir)1−x/C system on the electro-oxidation of methanol in acid media. J. Power Sources 2008, 179, 42–49. [Google Scholar] [CrossRef]
- Geng, D.S.; Matsuki, D.; Wang, J.J.; Kawaguchi, T.; Sugimoto, W.; Takasu, Y. Activity and Durability of Ternary PtRuIr/C for Methanol Electro-oxidation. J. Electrochem. Soc. 2009, 156, B397–B402. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Zhong, H.; Zhu, X.; Tian, Z.; Xu, D.; Yi, B. Preparation and characterization of carbon-supported PtRuIr catalyst with excellent CO-tolerant performance for proton-exchange membrane fuel cells. J. Catal. 2006, 238, 468–476. [Google Scholar] [CrossRef]
- Arico, A.S.; Bruce, P.; Scrosati, B.; Tarascon, J.-M.; van Schalkwijk, W. Nanostructured materials for advanced energy conversion and storage devices. Nat. Mater. 2005, 4, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Li, C.M. Nanostructured catalysts in fuel cells. J. Mater. Chem. 2011, 21, 4027–4036. [Google Scholar] [CrossRef]
- Loukrakpam, R.; Wanjala, B.N.; Yin, J.; Fang, B.; Luo, J.; Shao, M.; Protsailo, L.; Kawamura, T.; Chen, Y.; Petkov, V.; Zhong, C.-J. Structural and Electrocatalytic Properties of PtIrCo/C Catalysts for Oxygen Reduction Reaction. ACS Catal. 2011, 1, 562–572. [Google Scholar] [CrossRef]
- Cui, Z.; Kulesza, P.J.; Li, C.M.; Xing, W.; Jiang, S.P. Pd nanoparticles supported on HPMo-PDDA-MWCNT and their activity for formic acid oxidation reaction of fuel cells. Int. J. Hydrog. Energy 2011, 36, 8508–8517. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Wang, R.; Ji, S.; Wang, W.; Wang, Q.; Lei, Z. Amorphous CoSn alloys decorated by Pt as high efficiency electrocatalysts for ethanol oxidation. J. Power Sources 2011, 196, 8000–8003. [Google Scholar] [CrossRef]
- Zhang, L.; Xia, D. Electrocatalytic activity of ordered intermetallic PtSb for methanol electro-oxidation. Appl. Surf. Sci. 2006, 252, 2191–2195. [Google Scholar] [CrossRef]
- Youn, D.H.; Han, S.; Bae, G.; Lee, J.S. Carbon-supported PtPb intermetallic compounds for electrooxidation of methyl formate. Electrochem. Commun. 2011, 13, 806–809. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Li, H.; Wang, Q.; Kang, J.; Lei, Z. Facile synthesis of carbon-supported pseudo-core@shell PdCu@Pt nanoparticles for direct methanol fuel cells. Int. J. Hydrog. Energy 2011, 36, 839–848. [Google Scholar] [CrossRef]
- Wang, H.; Linkov, V.; Ji, S.; Zhang, W.; Lei, Z.; Wang, R. Highly Active, Carbon-supported, PdSn Nano-core, Partially Covered with Pt, as Catalysts for Methanol Oxidation. South Afr. J. Chem. 2012, 65, 69–74. [Google Scholar]
- Shrestha, S.; Liu, Y.; Mustain, W.E. Electrocatalytic Activity and Stability of Pt clusters on State-of-the-Art Supports: A Review. Catal. Rev. 2011, 53, 256–336. [Google Scholar] [CrossRef]
- Park, K.-W.; Choi, J.-H.; Kwon, B.-K.; Lee, S.-A.; Sung, Y.-E.; Ha, H.-Y.; Hong, S.-A.; Kim, H.; Wieckowski, A. Chemical and Electronic Effects of Ni in Pt/Ni and Pt/Ru/Ni Alloy Nanoparticles in Methanol Electrooxidation. J. Phys. Chem. B 2002, 106, 1869–1877. [Google Scholar] [CrossRef]
- Wang, R.; Jia, J.; Li, H.; Li, X.; Wang, H.; Chang, Y.; Kang, J.; Lei, Z. Nitrogen-doped carbon coated palygorskite as an efficient electrocatalyst support for oxygen reduction reaction. Electrochim. Acta 2011, 56, 4526–4531. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.; Ross, P.N.; Cairns, E.J. Methanol electrooxidation on well-characterized platinum-ruthenium bulk alloys. J. Phys. Chem. 1993, 97, 12020–12029. [Google Scholar] [CrossRef]
- Peng, Z.; Yang, H. Synthesis and Oxygen Reduction Electrocatalytic Property of Pt-on-Pd Bimetallic Heteronanostructures. J. Am. Chem. Soc. 2009, 131, 7542–7543. [Google Scholar] [CrossRef] [PubMed]
- Prabhuram, J.; Zhao, T.S.; Tang, Z.K.; Chen, R.; Liang, Z.X. Multiwalled Carbon Nanotube Supported PtRu for the Anode of Direct Methanol Fuel Cells. J. Phys. Chem. B 2006, 110, 5245–5252. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, H.; Peng, F.; Liang, J.; Yu, H.; Yang, J. MnO2/CNT Supported Pt and PtRu Nanocatalysts for Direct Methanol Fuel Cells. Langmuir 2009, 25, 7711–7717. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.; Gouws, S.; Ferg, E. Characterization of Pt Catalysts for PEM Fuel Cells. Mol. Cryst. Liquid Cryst. 2012, 555, 149–157. [Google Scholar] [CrossRef]
- Schulenburg, H.; Durst, J.; Müller, E.; Wokaun, A.; Scherer, G.G. Real surface area measurements of Pt3Co/C catalysts. J. Electroanal. Chem. 2010, 642, 52–60. [Google Scholar] [CrossRef]
- Jiang, J.; Kucernak, A. Electrooxidation of small organic molecules on mesoporous precious metal catalysts. I: CO and methanol on platinum. J. Electroanal. Chem. 2002, 533, 153–165. [Google Scholar] [CrossRef]
- Jiang, J.; Kucernak, A. Electrooxidation of small organic molecules on mesoporous precious metal catalysts. II: CO and methanol on platinum–ruthenium alloy. J. Electroanal. Chem. 2003, 543, 187–199. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, Y.; Wang, R.; Wang, H.; Liao, S.; Key, J.; Linkov, V.; Ji, S. The Effect of PtRuIr Nanoparticle Crystallinity in Electrocatalytic Methanol Oxidation. Materials 2013, 6, 1621-1631. https://doi.org/10.3390/ma6051621
Ma Y, Wang R, Wang H, Liao S, Key J, Linkov V, Ji S. The Effect of PtRuIr Nanoparticle Crystallinity in Electrocatalytic Methanol Oxidation. Materials. 2013; 6(5):1621-1631. https://doi.org/10.3390/ma6051621
Chicago/Turabian StyleMa, Yanjiao, Rongfang Wang, Hui Wang, Shijun Liao, Julian Key, Vladimir Linkov, and Shan Ji. 2013. "The Effect of PtRuIr Nanoparticle Crystallinity in Electrocatalytic Methanol Oxidation" Materials 6, no. 5: 1621-1631. https://doi.org/10.3390/ma6051621
APA StyleMa, Y., Wang, R., Wang, H., Liao, S., Key, J., Linkov, V., & Ji, S. (2013). The Effect of PtRuIr Nanoparticle Crystallinity in Electrocatalytic Methanol Oxidation. Materials, 6(5), 1621-1631. https://doi.org/10.3390/ma6051621