Density Functional Theory (DFT) Study of Coumarin-based Dyes Adsorbed on TiO2 Nanoclusters—Applications to Dye-Sensitized Solar Cells

 and
and
Abstract
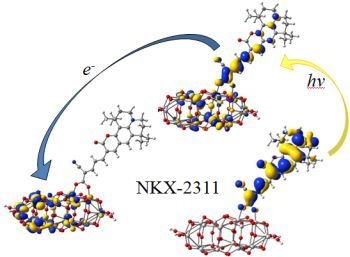
:
1. Introduction
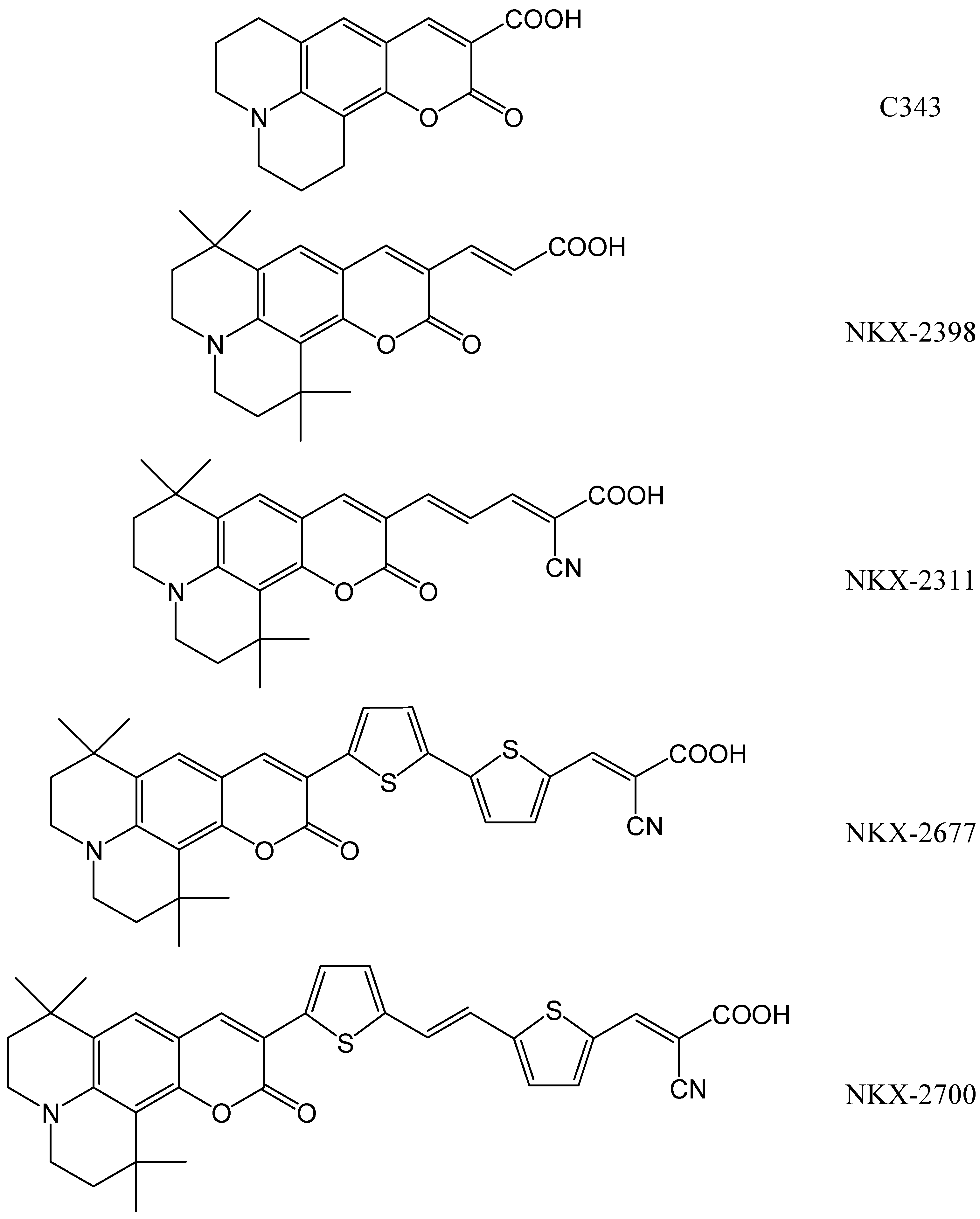
- (i)
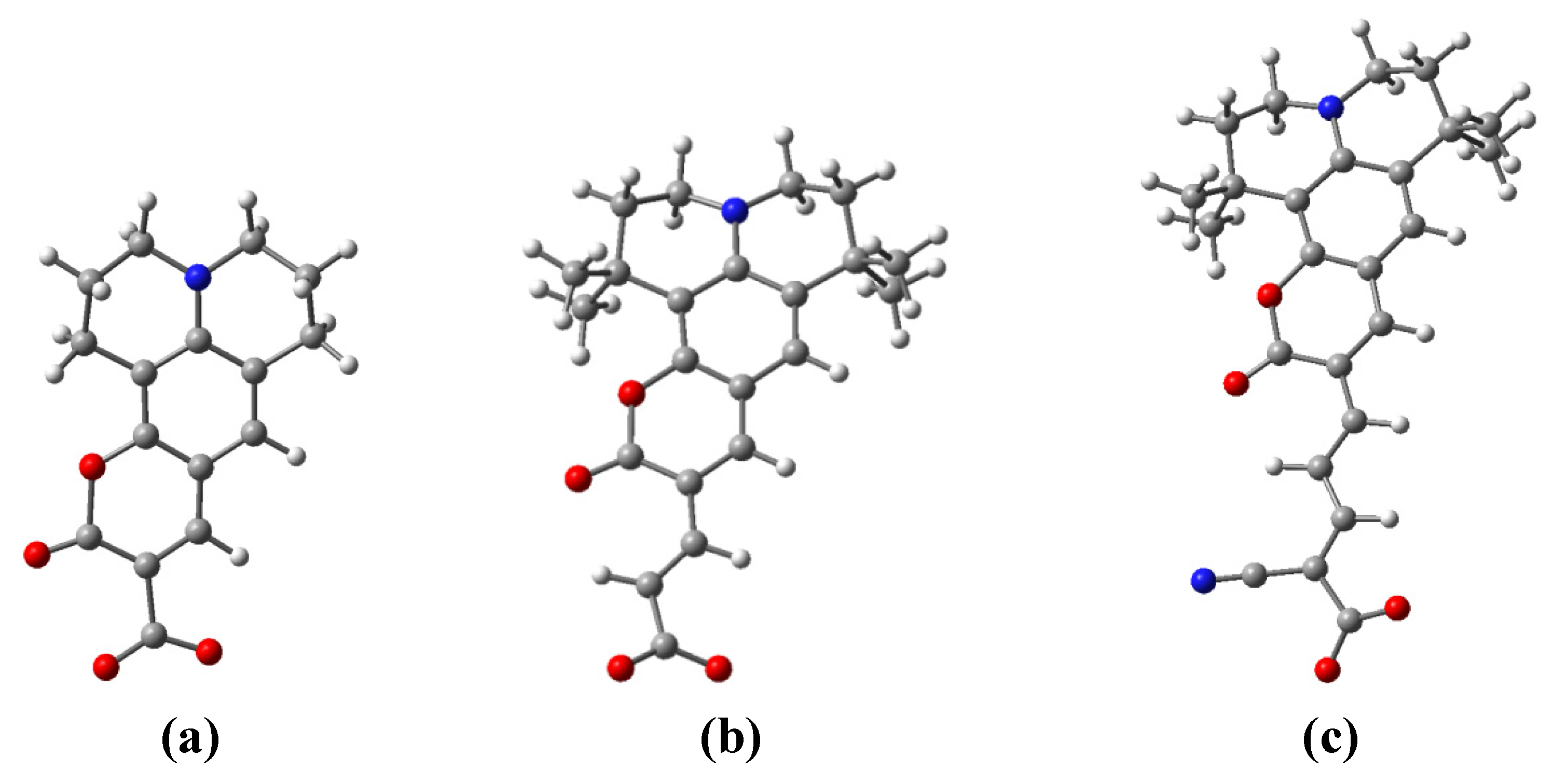
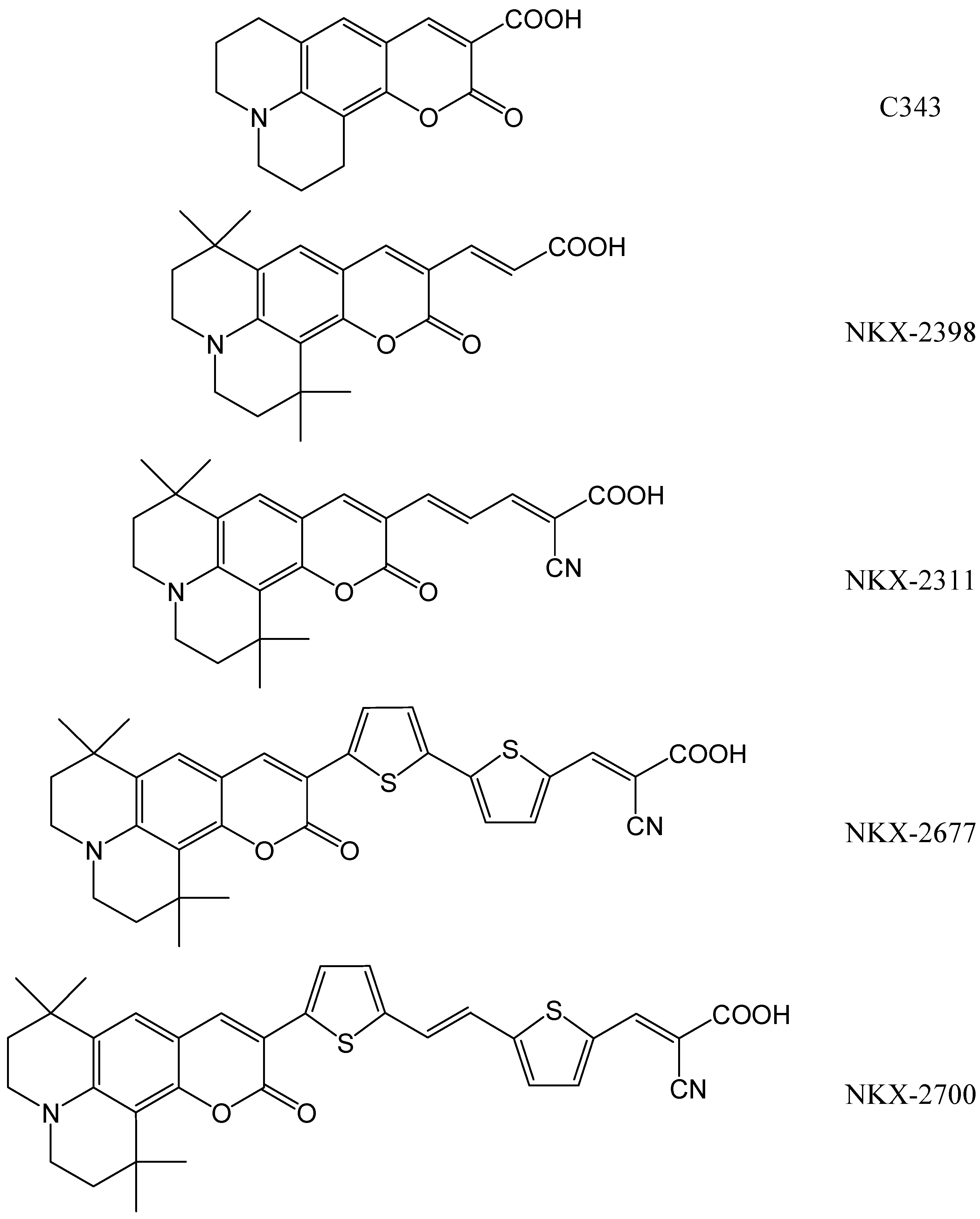
- 1-oxo-2,3,6,7-tetrahydro-1H,5H,11H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid (C343),
- (ii)
- 3-(1,1,6,6-tetramethyl-10-oxo-2,3,5,6-tetrahydro-1H,4H,10H-11-oxa-3a-azabenzo[de]anthracen-9-yl)-acrylic acid (NKX-2398) and
- (iii)
- 2-cyano-5-(1,1,6,6-tetramethyl-10-oxo-2,3,5,6-tetrahydro-1H,4H,10H-11-oxa-3a-aza-benzo[de]anthracen-9-yl)-penta-2,4-dienoic acid (NKX-2311).
2. Computational Details
3. Results and Discussion
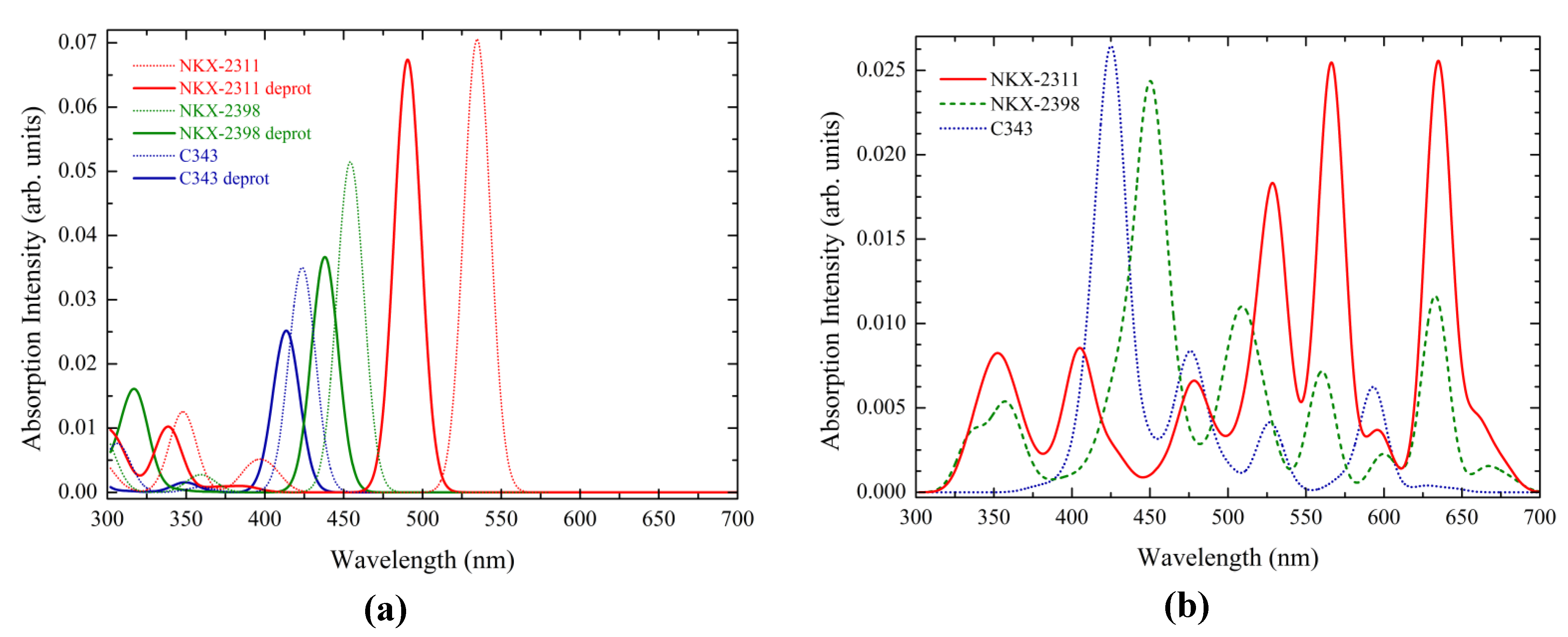
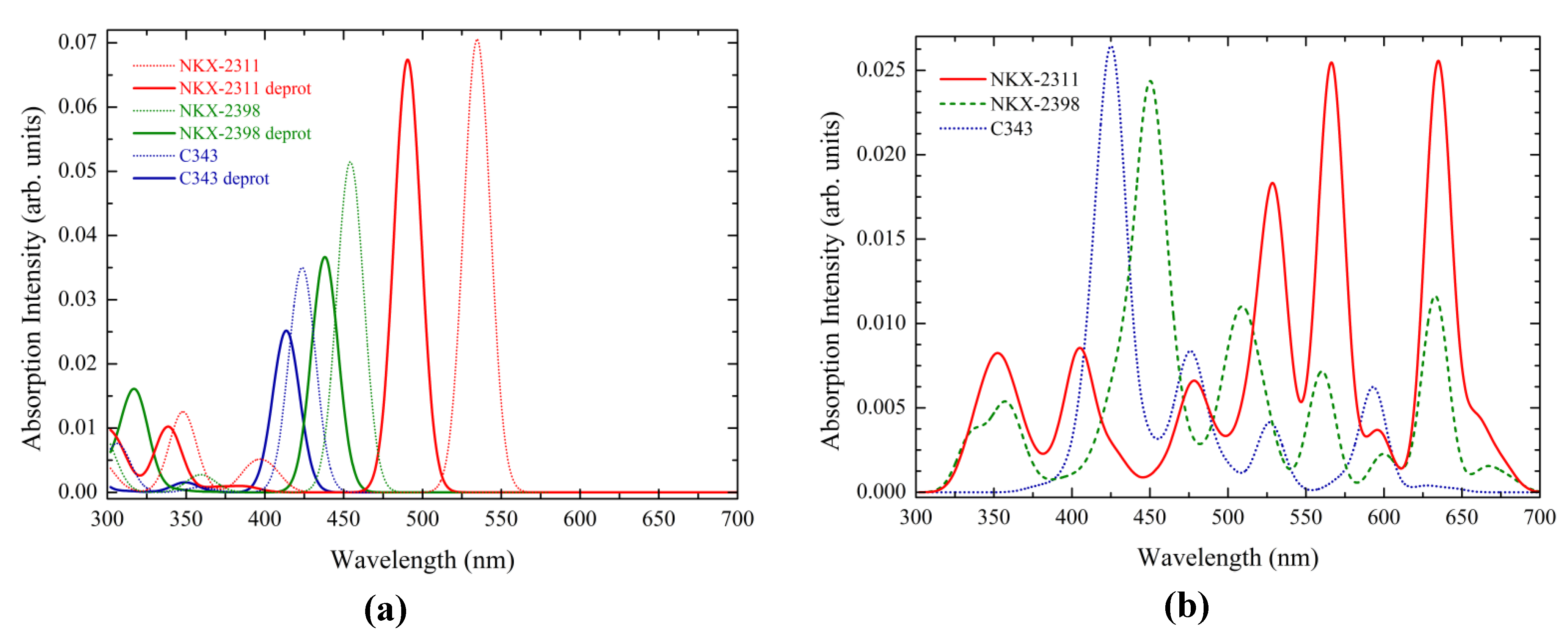
3.1. Absorption Spectrum
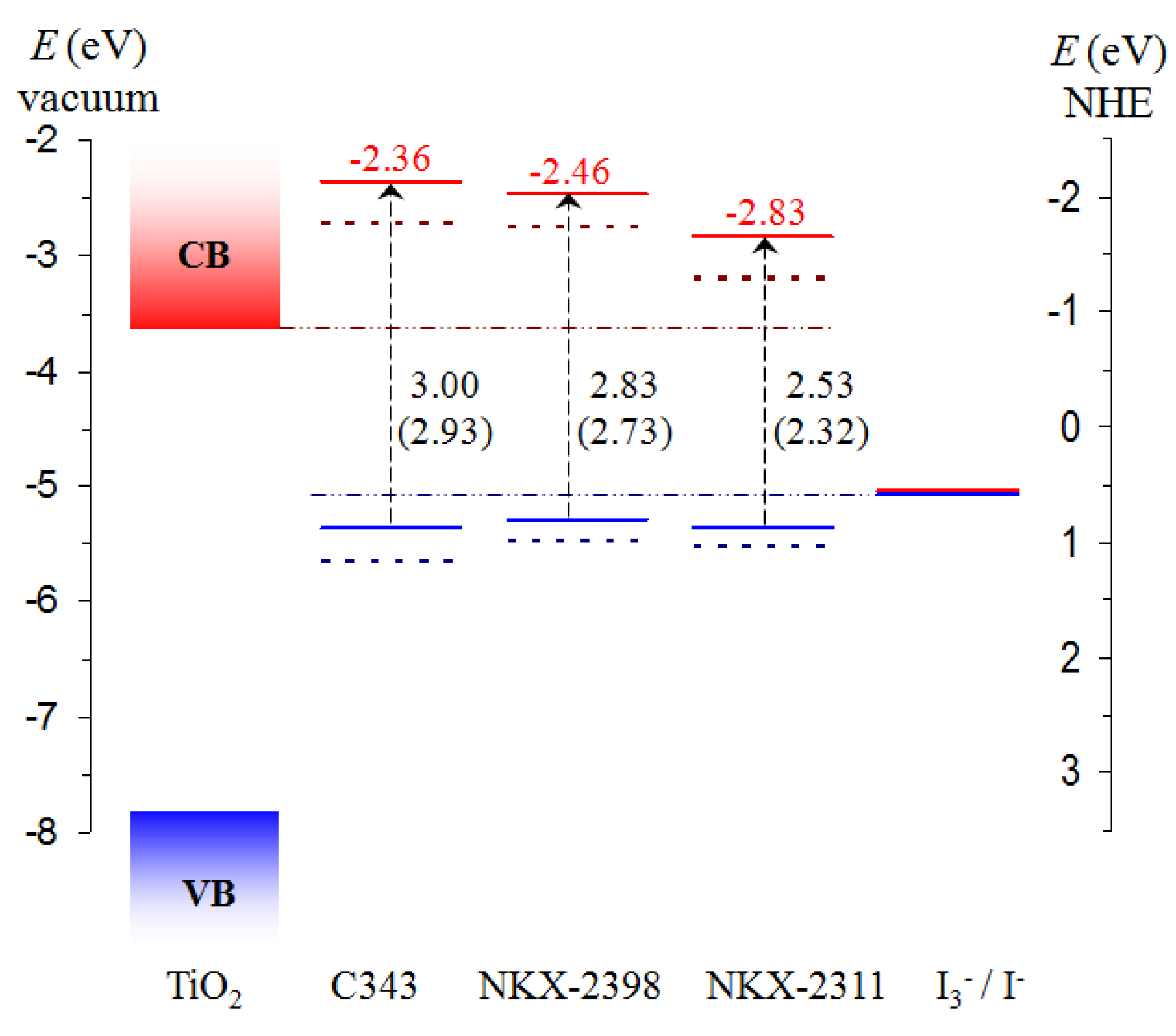
3.2. Energy Level Alignment and Electron Transfer
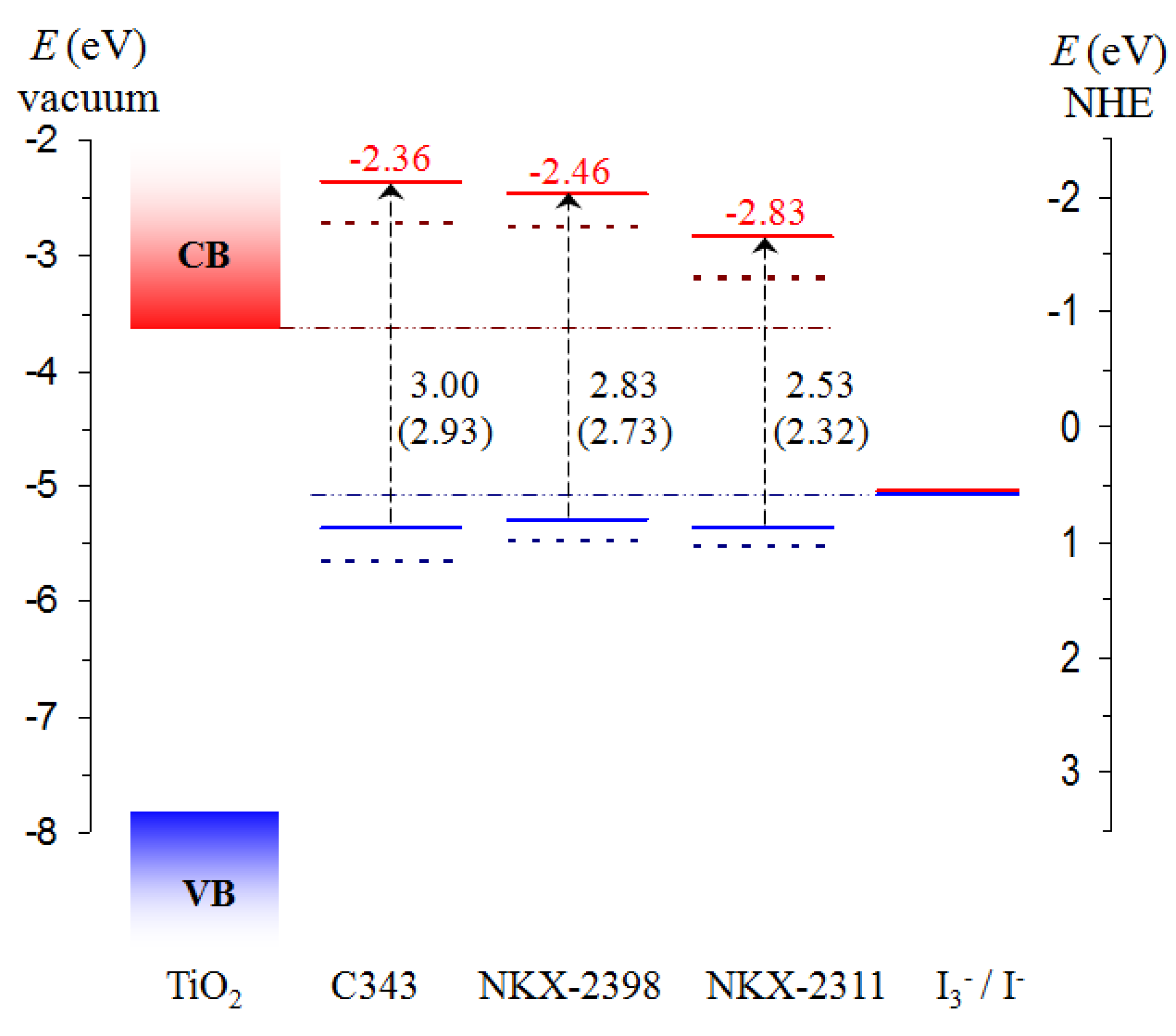
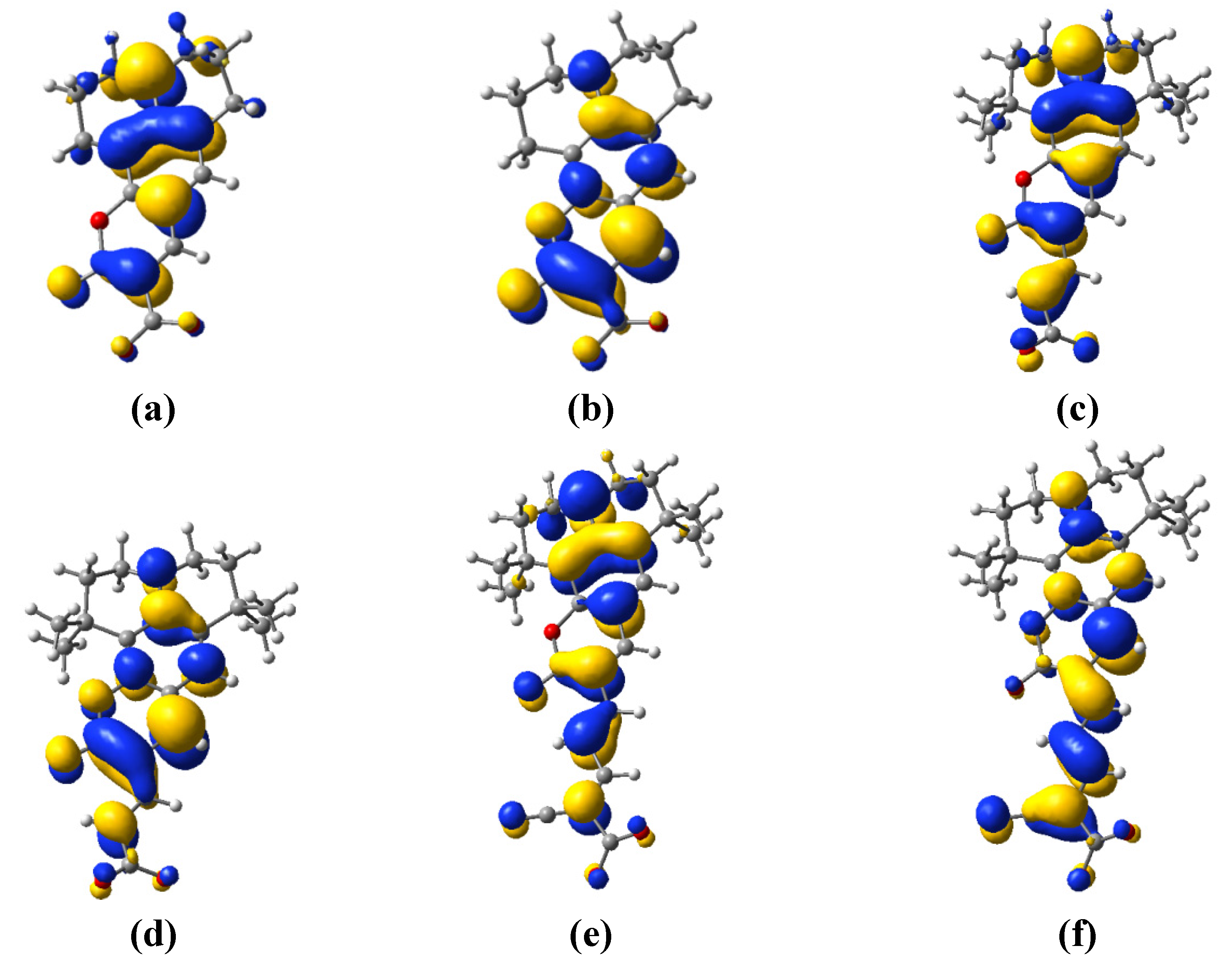
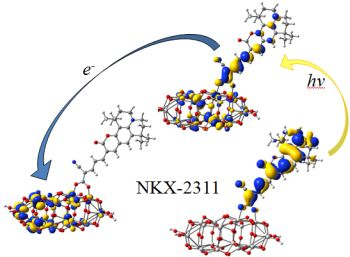
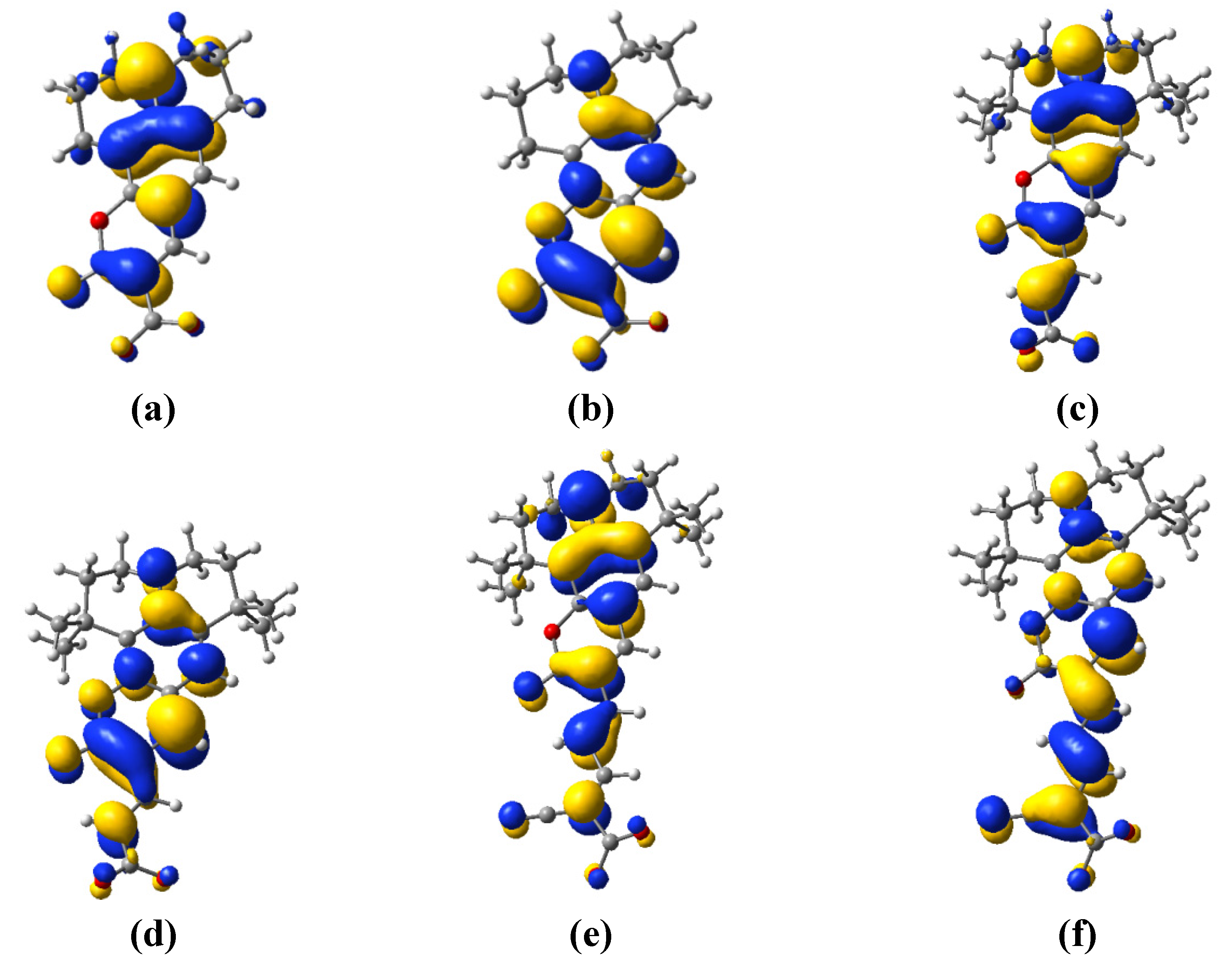
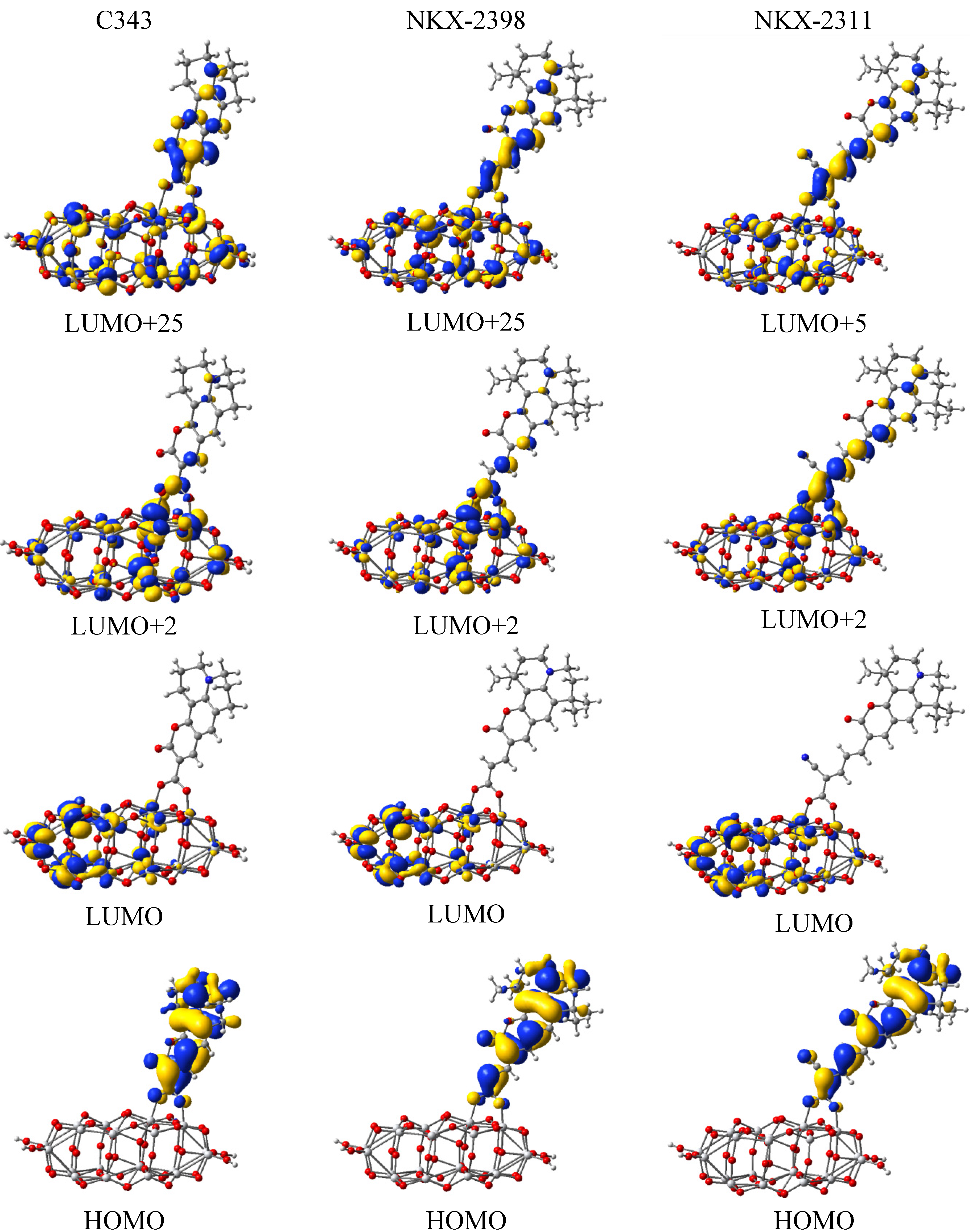
3.3. Electron Transfer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | MO | Contribution to electron density of the donor unit (%) | Contribution to electron density of the acceptor unit (%) | Total (%) |
|---|---|---|---|---|
| C343 | HOMO | 98.473 | 1.527 | 100 |
| LUMO | 97.572 | 2.428 | 100 | |
| NKX-2398 | HOMO | 85.845 | 14.155 | 100 |
| LUMO | 90.806 | 9.194 | 100 | |
| NKX-2311 | HOMO | 79.586 | 20.414 | 100 |
| LUMO | 57.181 | 42.819 | 100 |
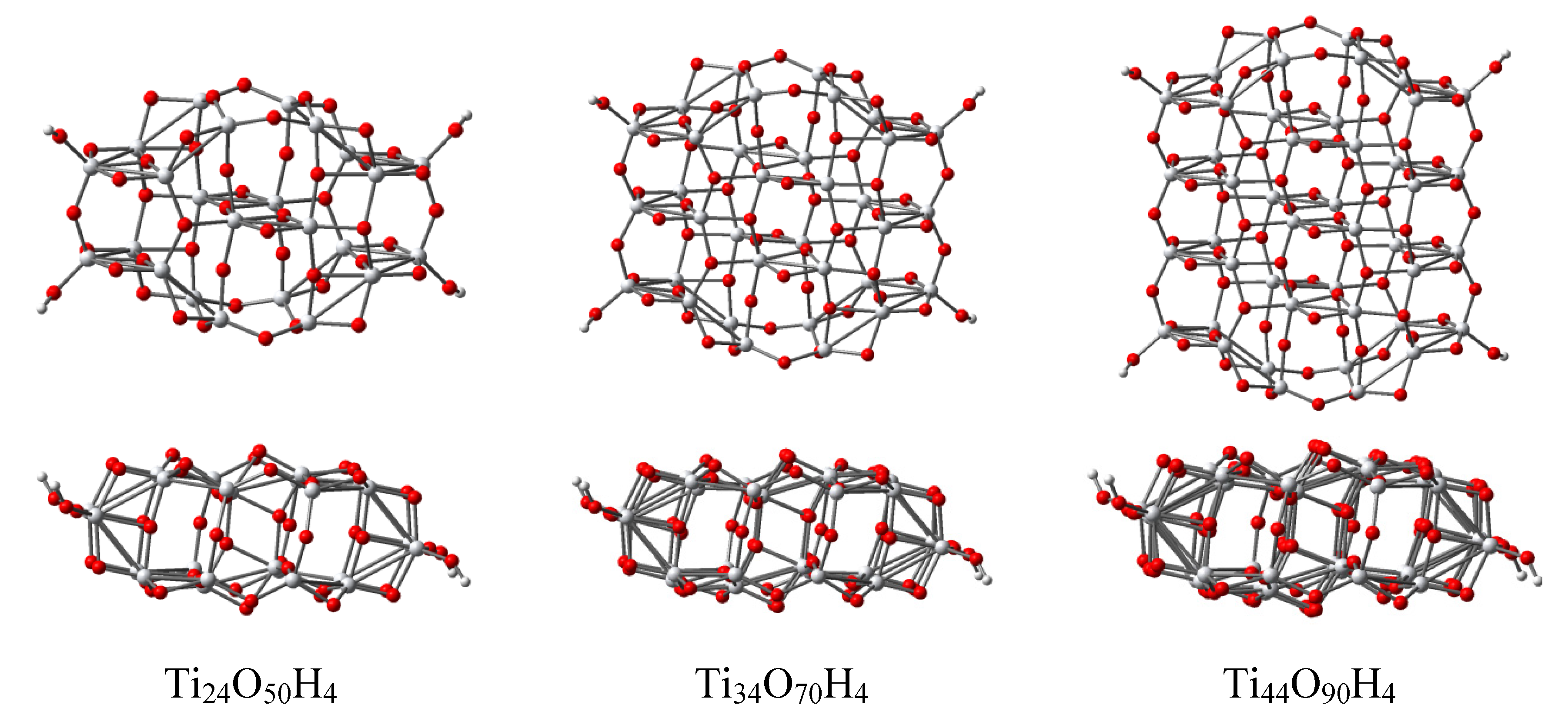
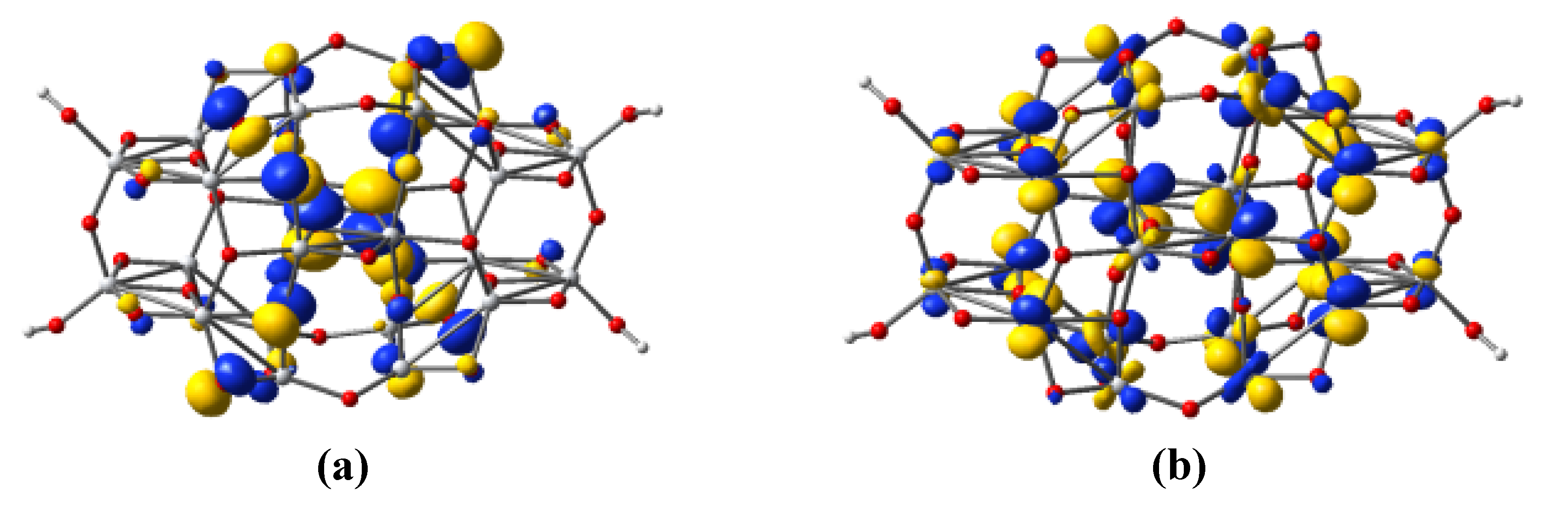
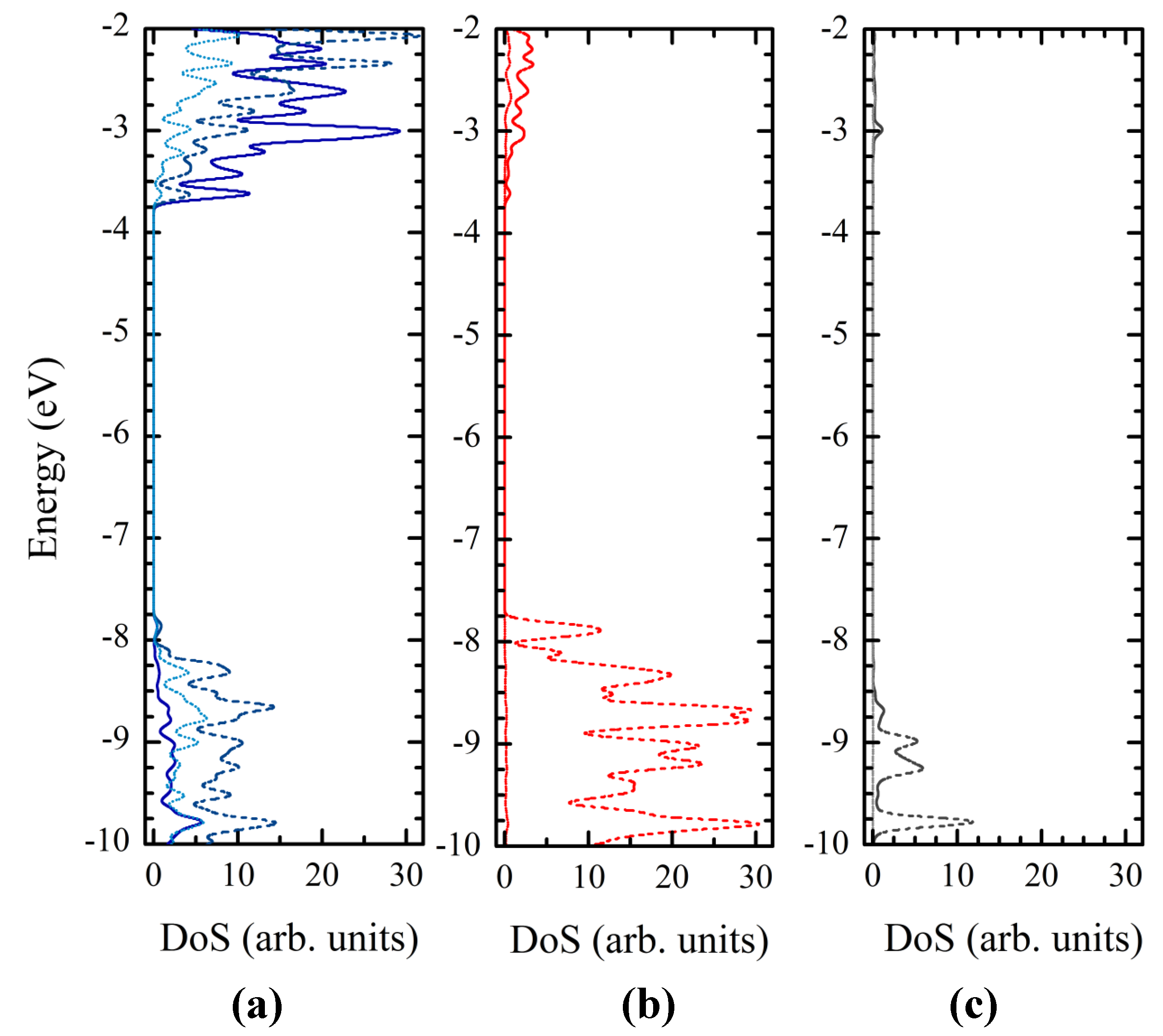
3.4. Titania Nanoclusters
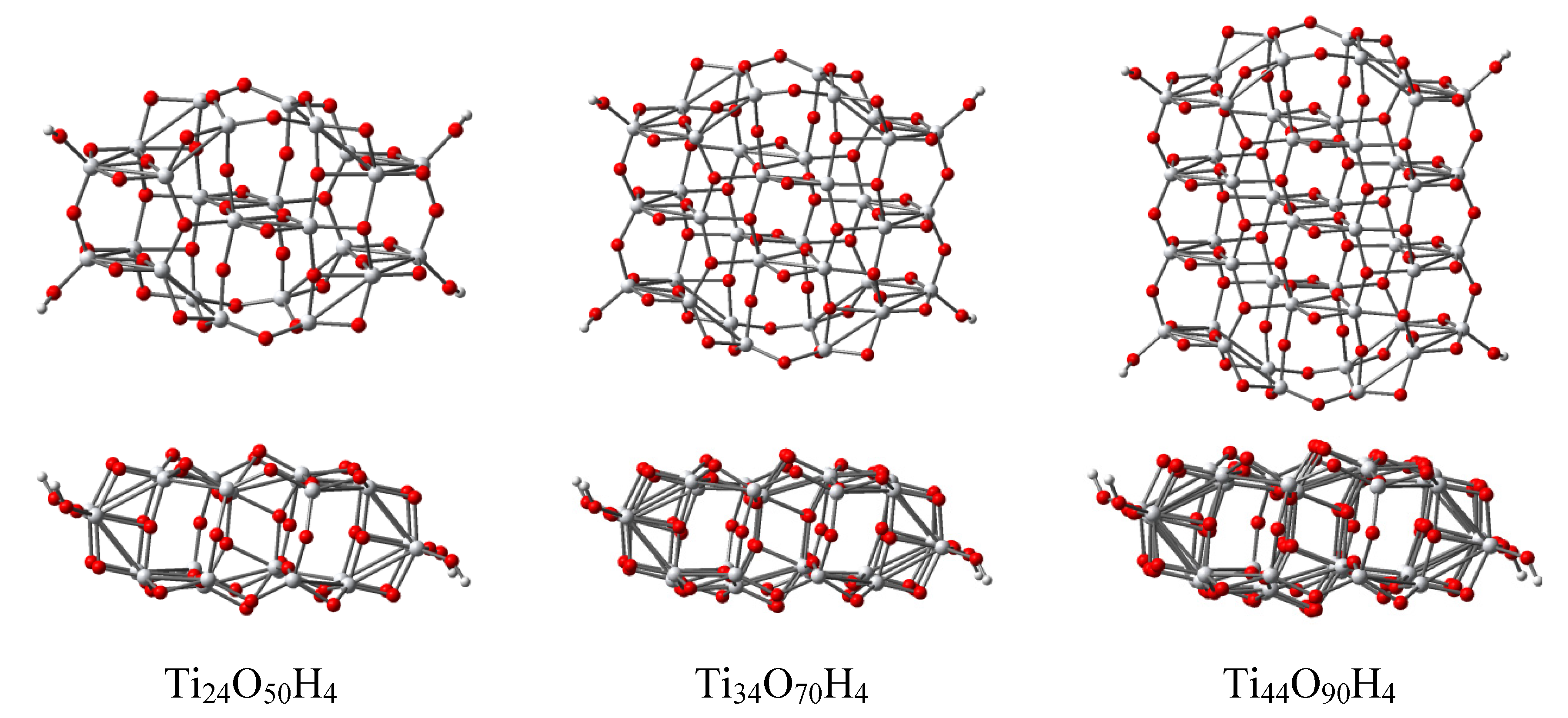
| Parameter | Ti24O50H4 | Ti34O70H4 | Ti44O90H4 | TiO2 (bulk) |
|---|---|---|---|---|
| r(Ti–O) | 1.909 | 1.908 | 1.916 | 1.950 |
| σr(Ti–O) | 0.133 | 0.101 | 0.105 | 0.022 |
| Length | 12.76 | 12.54 | 12.50 | 12.04 |
| Width | 7.39 | 10.91 | 14.34 | – |
| Width (bulk) | 7.59 | 11.37 | 15.14 | – |
| Basis set | Ti24O50H4 | Ti34O70H4 | Ti44O90H4 | (TiO2)38 [16] | ||||
|---|---|---|---|---|---|---|---|---|
| ECB | CBOP | ECB | CBOP | ECB | CBOP | ECB | CBOP | |
| 3-21G* | −3.20 | −3.98 | −3.41 | −4.12 | −3.51 | −4.14 | −2.77 | −3.35 |
| DZVP | −3.86 | −4.63 | −3.98 | −4.67 | −4.02 | −4.69 | −3.44 | −4.04 |
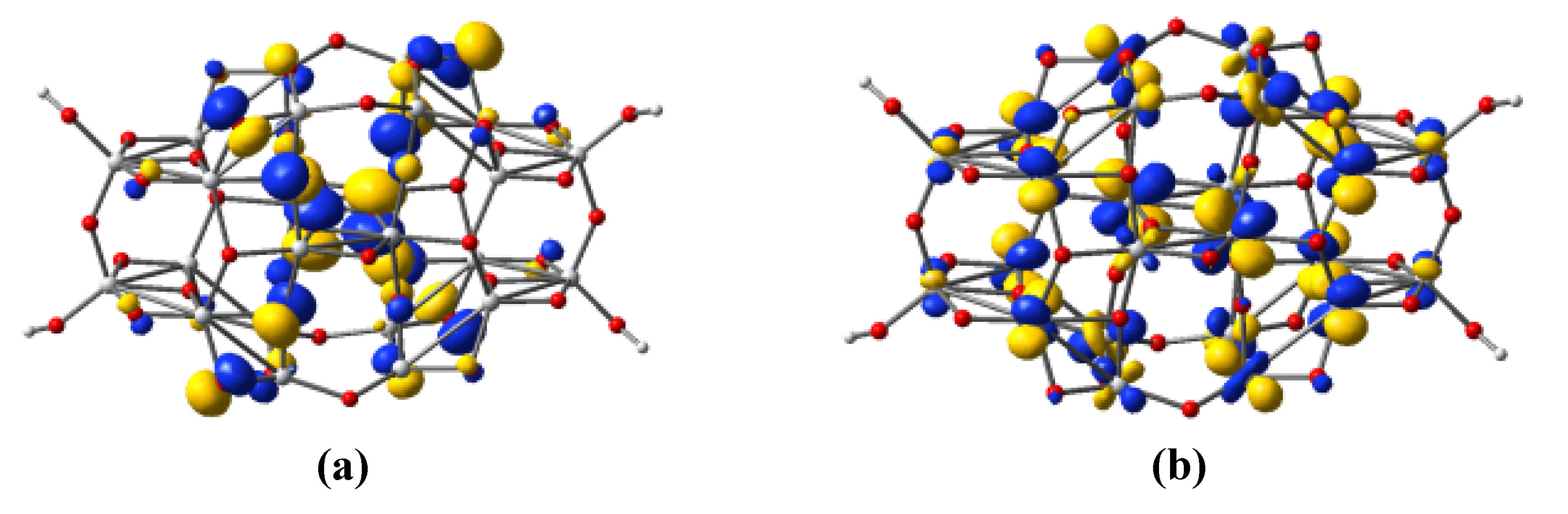
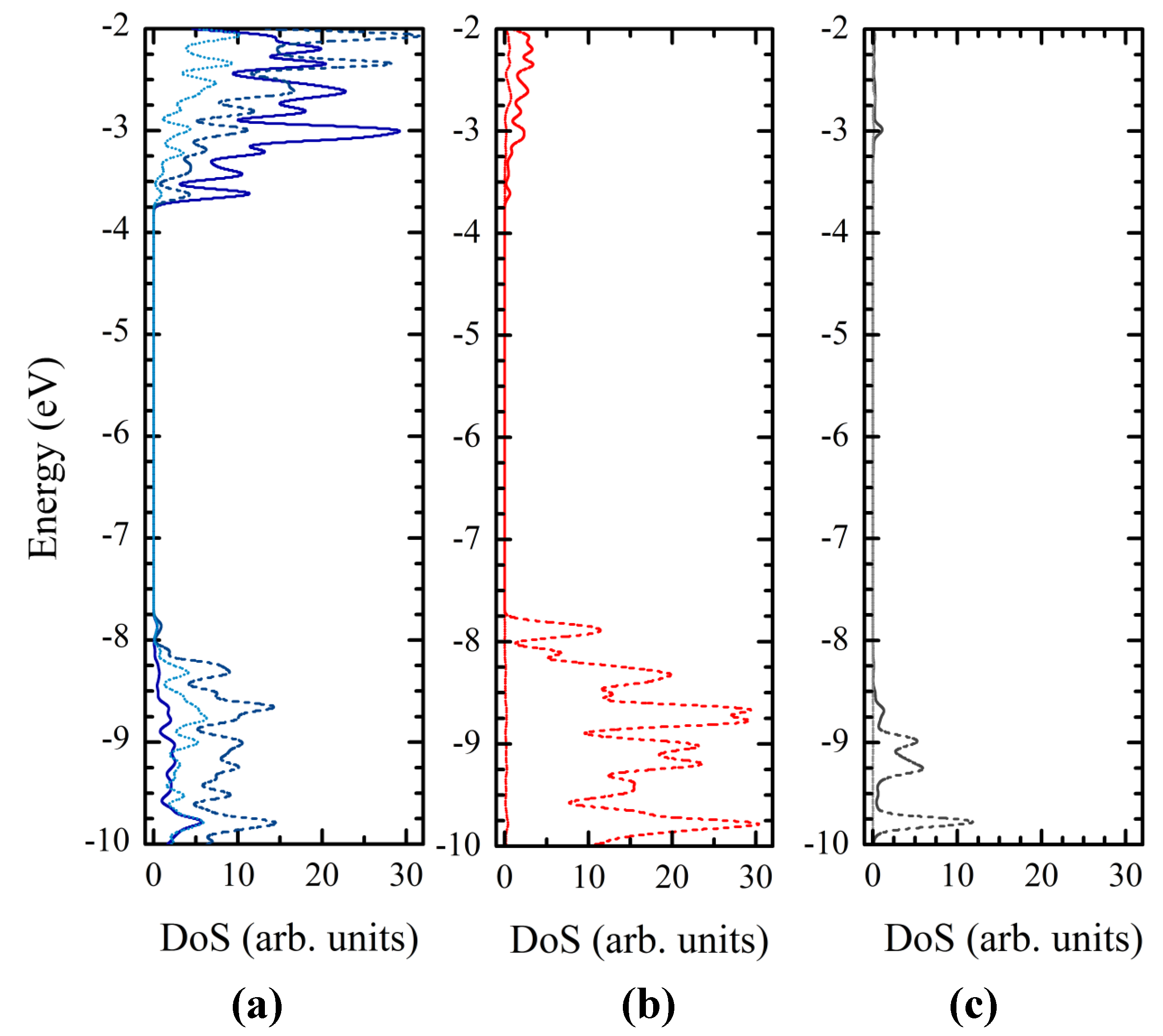
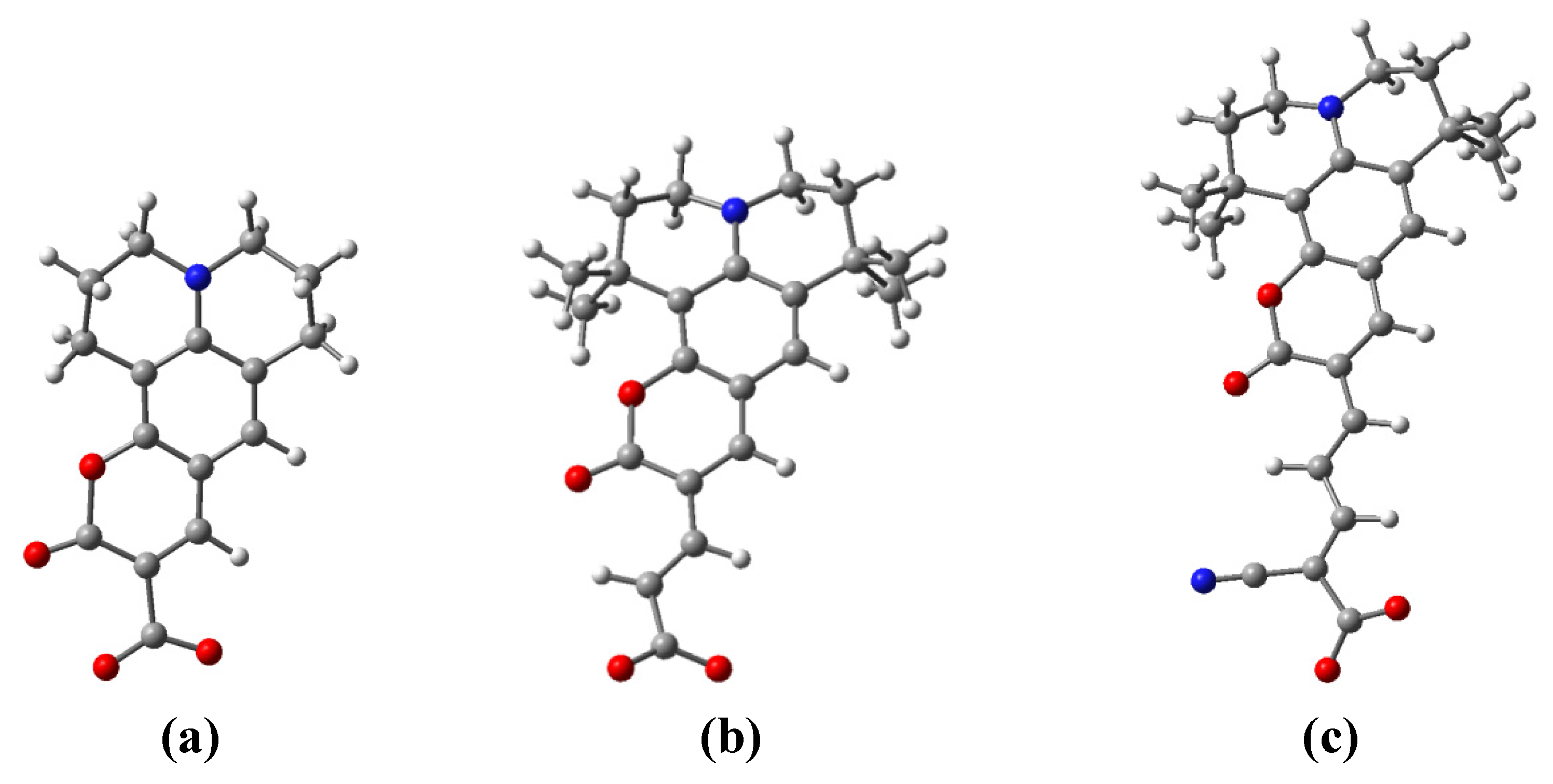
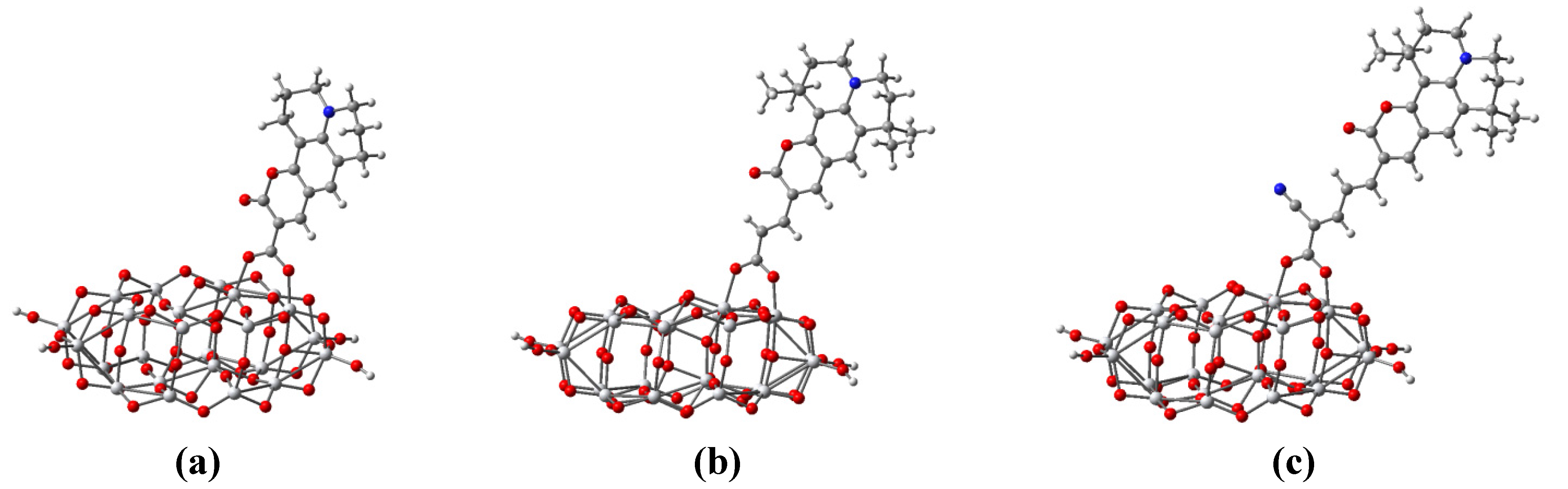
3.5. Dye Adsorption
| Parameter | C343-Ti24O50H4 | NKX-2398-Ti24O50H4 | NKX-2311-Ti24O50H4 |
|---|---|---|---|
| r(Ti–O1) | 1.977 | 1.969 | 1.986 |
| r(Ti–O2) | 2.128 | 2.115 | 2.138 |
| θ | 105.2 | 113.5 | 122.2 |
| τ | 16.5 | 23.5 | 32.6 |
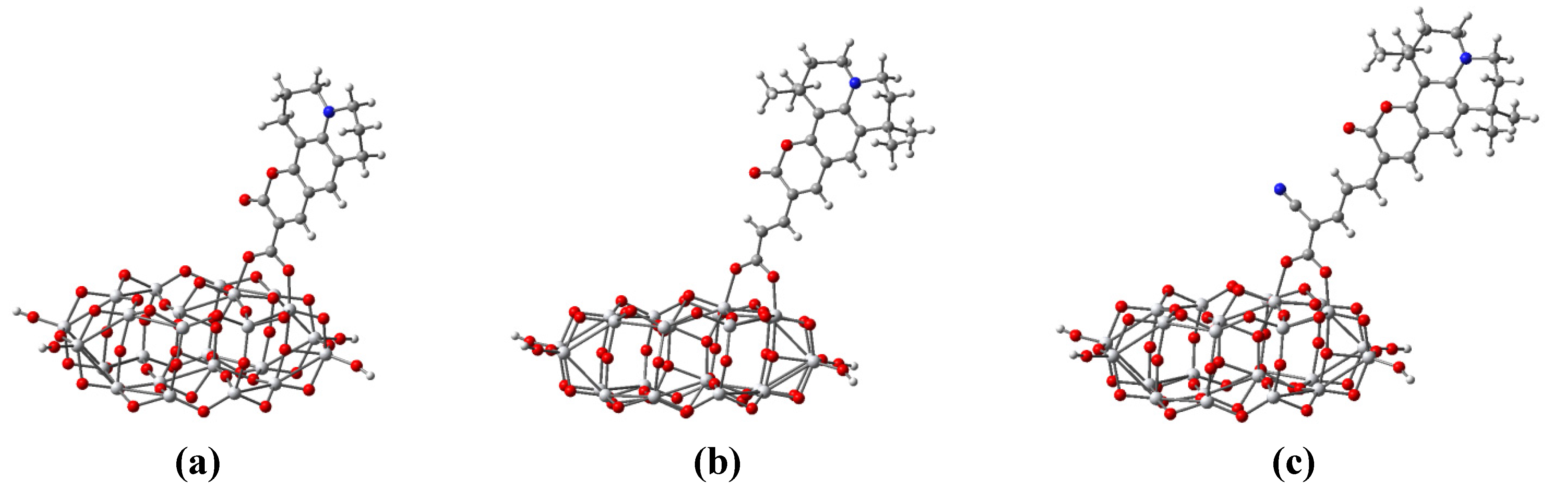
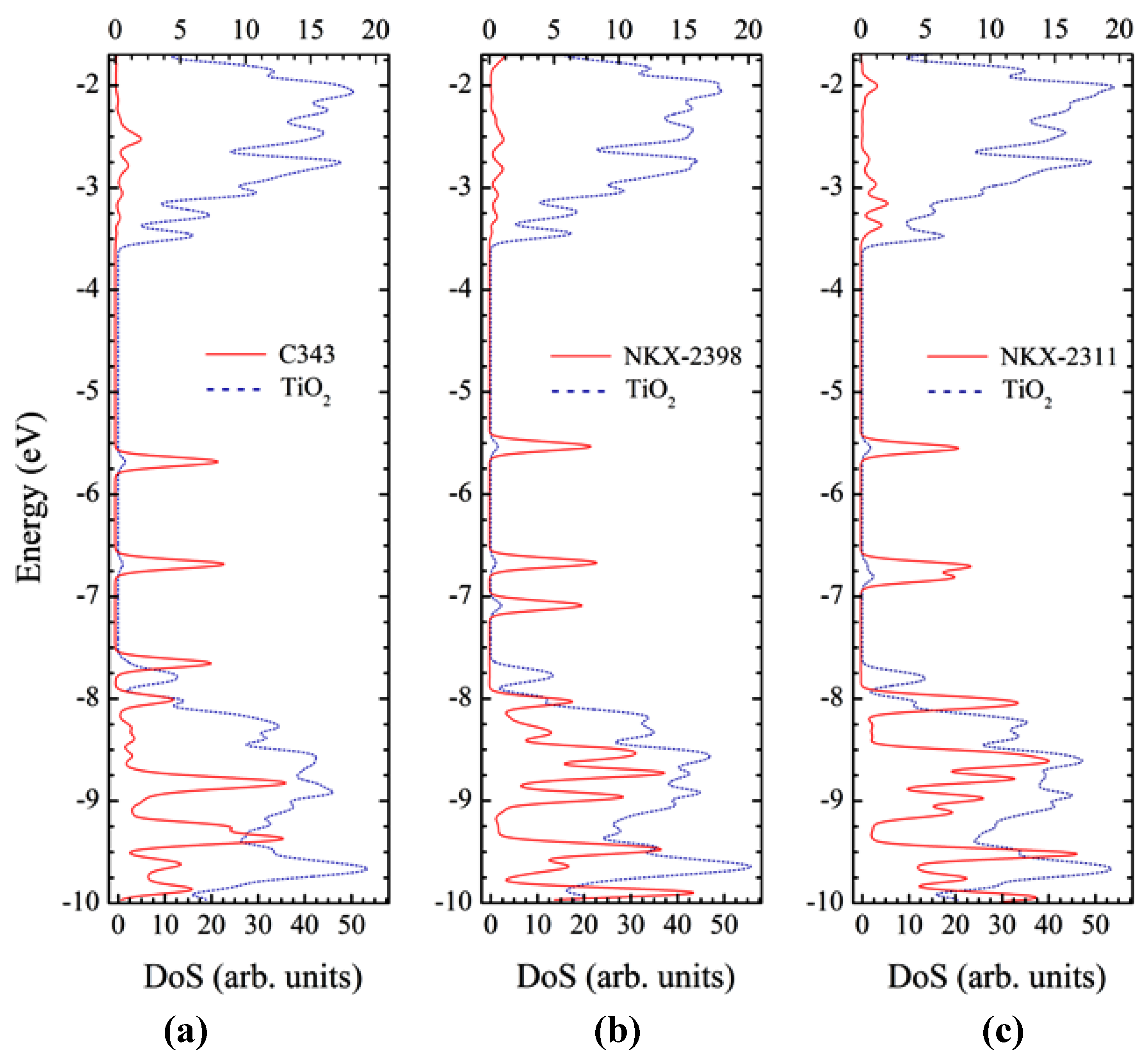
3.6. Optical Properties of Adsorbed Dyes
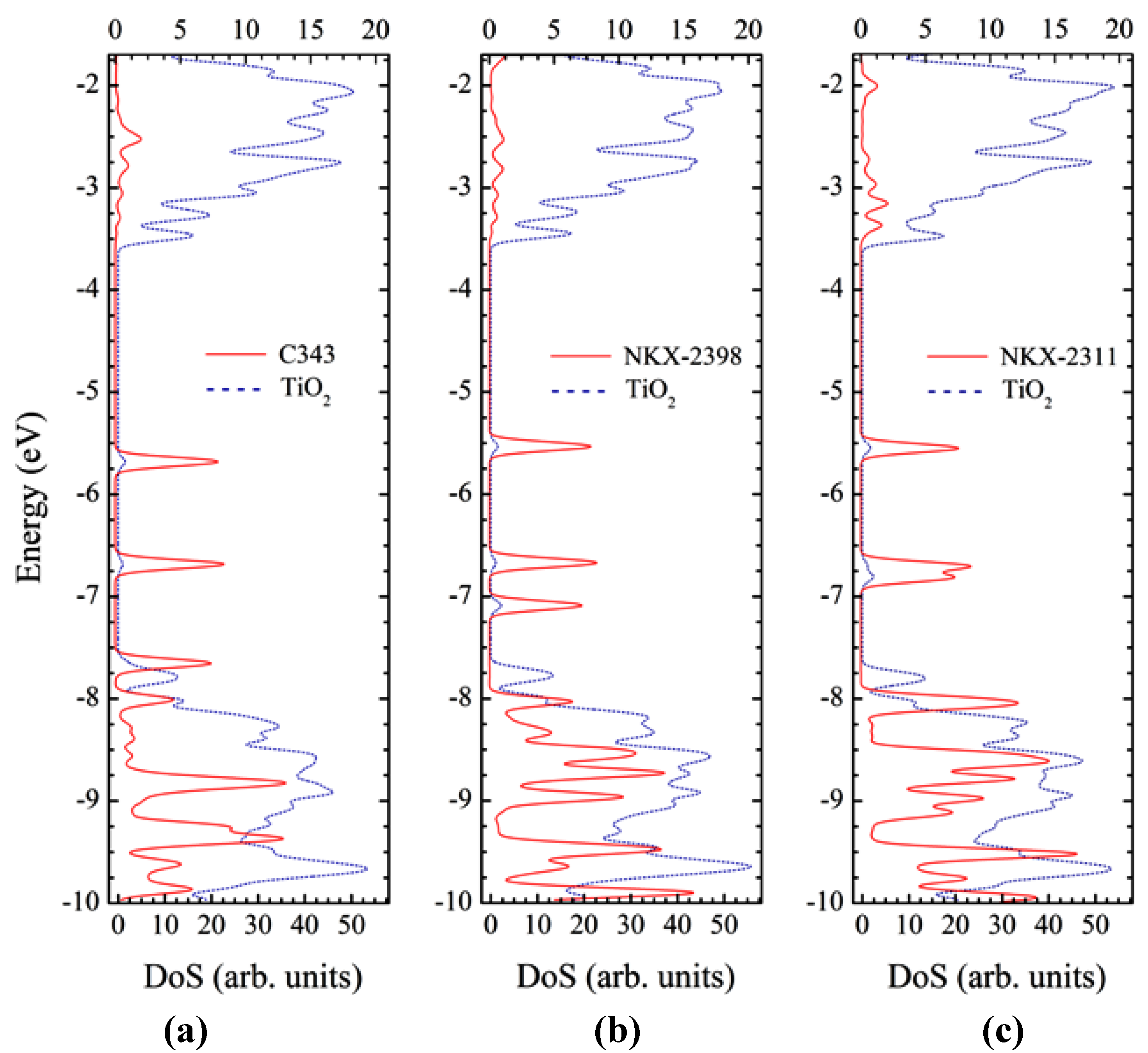

4. Conclusions
Acknowledgments
References
- Gratzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar]
- Ahn, K.S.; Yoo, S.J.; Kang, M.S.; Lee, J.W.; Sung, Y.E. Tandem dye-sensitized solar cell-powered electrochromic devices for the photovoltaic-powered smart window. J. Power Sources 2007, 168, 533–536. [Google Scholar]
- Baetens, R.; Jelle, B.P.; Gustavsen, A. Properties, requirements and possibilities of smart windows for dynamic daylight and solar energy control in buildings: A state-of-the-art review. Sol. Energy Mater. Sol. Cells 2010, 94, 87–105. [Google Scholar]
- Nazeeruddin, M.K.; de Angelis, F.; Fantacci, S.; Selloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Bessho, M.; Gratzel, T. Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers. J. Am. Chem. Soc. 2005, 127, 16835–16846. [Google Scholar]
- Peter, L.M. The gratzel cell: Where next? J. Phys. Chem. Lett. 2011, 2, 1861–1867. [Google Scholar]
- Hardin, B.E.; Snaith, H.J.; McGehee, M.D. The renaissance of dye-sensitized solar cells. Nat. Photon. 2012, 6, 162–169. [Google Scholar]
- Yella, A.; Lee, H.W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.G.; Yeh, C.Y.; Zakeeruddin, S.M.; Gratzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)-based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar]
- Ito, S.; Zakeeruddin, S.M.; Humphry-Baker, R.; Liska, P.; Charvet, R.; Comte, P.; Nazeeruddin, M.K.; Pechy, P.; Takata, M.; Miura, H.; et al. High-efficiency organic-dye-sensitized solar cells controlled by nanocrystalline-TiO2 electrode thickness. Adv. Mater. 2006, 18, 1202–1205. [Google Scholar]
- Hwang, S.; Lee, J.H.; Park, C.; Lee, H.; Kim, C.; Park, C.; Lee, M.H.; Lee, W.; Park, J.; Kim, K.; Park, C.; Kim, N.G. A highly efficient organic sensitizer for dye-sensitized solar cells. Chem. Commun. 2007, 4887–4889. [Google Scholar]
- Mishra, A.; Fischer, M.K.R.; Bauerle, P. Metal-free organic dyes for dye-sensitized solar cells: From structure: Property relationships to design rules. Angew. Chem. Int. Ed. 2009, 48, 2474–2499. [Google Scholar]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar]
- Hamann, T.W.; Jensen, R.A.; Martinson, A.B.F.; van Ryswyk, H.; Hupp, J.T. Advancing beyond current generation dye-sensitized solar cells. Energy Environ. Sci. 2008, 1, 66–78. [Google Scholar]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Grätzel, M.; Nazeeruddin, M.K. Influence of the sensitizer adsorption mode on the open-circuit potential of dye-sensitized solar cells. Nano Lett. 2007, 7, 3189–3195. [Google Scholar]
- Hagberg, D.P.; Yum, J.H.; Lee, H.J.; de Angelis, F.; Marinado, T.; Martin Karlsson, K.; Humphry-Baker, R.; Sun, L.; Hagfeldt, A.; Gratzel, M.; et al. Molecular engineering of organic sensitized for dye-sensitized solar cells applications. J. Am. Chem. Soc. 2008, 130, 6259–6266. [Google Scholar]
- De Angelis, F.; Fantacci, S.; Sgamellotti, A. An integrated computational tool for the study of the optical properties of nanoscale devices: Application to solar cells and molecular wires. Theor. Chem. Acc. 2007, 117, 1093–1104. [Google Scholar]
- De Angelis, F.; Fantacci, S.; Selloni, A. Alignment of the dye’s molecular levels with the TiO(2) band edges in dye-sensitized solar cells: A DFT-TDDFT study. Nanotechnology 2008, 19. [Google Scholar] [CrossRef]
- Seo, K.D.; Song, H.M.; Lee, M.J.; Pastore, M.; Anselmi, C.; de Angelis, F.; Nazeeruddin, M.K.; Gräetzel, M.; Kim, H.K. Coumarin dyes containing low-band-gap chromophores for dye-sensitised solar cells. Dyes Pigment. 2011, 90, 304–310. [Google Scholar]
- Hara, K.; Sayama, K.; Arakawa, H.; Ohga, Y.; Shinpo, A.; Suga, S. A coumarin-derivative dye sensitized nanocrystalline TiO2 solar cell having a high solar-energy conversion efficiency up to 5.6%. Chem. Commun. 2001, 569–570. [Google Scholar]
- Hara, K.; Miyamoto, K.; Abe, Y.; Yanagida, M. Electron transport in coumarin-dye-sensitized nanocrystalline TiO2 electrodes. J. Phys. Chem. B 2005, 109, 23776–23778. [Google Scholar]
- Wang, Z.S.; Cui, Y.; Hara, K.; Dan-Oh, Y.; Kasada, C.; Shinpo, A. A High-light-harvesting-efficiency coumarin dye for stable dye-sensitized solar cells. Adv. Mater. 2007, 19, 1138–1141. [Google Scholar]
- Wang, Z.S.; Cui, Y.; Dan-oh, Y.; Kasada, C.; Shinpo, A.; Hara, K. Thiophene-functionalized coumarin dye for efficient dye-sensitized solar cells: Electron lifetime improved by coadsorption of deoxycholic acid. J. Phys. Chem. C 2007, 111, 7224–7230. [Google Scholar]
- Ooyama, Y.; Harima, Y. Molecular designs and syntheses of organic dyes for dye-sensitized solar cells. Eur. J. Org. Chem. 2009, 2009, 2903–2934. [Google Scholar]
- Hara, K.; Sato, T.; Katoh, R.; Furube, A.; Ohga, Y.; Shinpo, A.; Suga, S.; Sayama, K.; Sugihara, H.; Arakawa, H. Molecular design of coumarin dyes for efficient dye-sensitized solar cells. J. Phys. Chem. B 2003, 107, 597–606. [Google Scholar]
- Kurashige, Y.; Nakajima, T.; Kurashige, S.; Hirao, K. Theoretical investigation of the excited states of coumarin dyes for dye-sensitized solar cells. J. Phys. Chem. A 2007, 111, 5544–5548. [Google Scholar]
- Preat, J.; Loos, P.-F.; Assfeld, X.; Jacquemin, D.; Perpète, E.A. A TD-DFT investigation of UV spectra of pyranoïdic dyes: A NCM vs. PCM comparison. J. Mol. Struct. Theochem. 2007, 808, 85–91. [Google Scholar]
- Zhang, X.; Zhang, J.-J.; Xia, Y.-Y. Molecular design of coumarin dyes with high efficiency in dye- sensitized solar cells. J. Photochem. Photobiol. A Chem. 2008, 194, 167–172. [Google Scholar]
- Sanchez-de-Armas, R.; San Miguel, M.A.; Oviedo, J.; Sanz, J.F. Coumarin derivatives for dye sensitized solar cells: A TD-DFT study. Phys. Chem. Chem. Phys. 2012, 14, 225–233. [Google Scholar]
- Kondov, I.; Wang, H.; Thoss, M. Computational study of titanium (IV) complexes with organic chromophores. Int. J. Quantum Chem. 2006, 106, 1291–1303. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, B37, 785–789. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–311. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent Density-Functional Response theory: Characterization and correlation of the time dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03 Citation. Avaiable online: http://www.gaussian.com/g_misc/g03/citation_g03.htm (accessed on 18 April 2013).
- Badaeva, E.; Albert, V.V.; Kilina, S.; Koposov, A.; Sykora, M.; Tretiak, S. Effect of deprotonation on absorption and emission spectra of Ru(II)-bpy complexes functionalized with carboxyl groups. Phys. Chem. Chem. Phys. 2010, 12, 8902–8913. [Google Scholar]
- Fantacci, S.; de Angelis, F.; Selloni, A. Absorption spectrum and solvatochromism of the [Ru(4,4′-COOH-2,2′-bpy)2(NCS)2] molecular dye by time dependent density functional theory. J. Am. Chem. Soc. 2003, 125, 4381–4387. [Google Scholar]
- Gueymard, C. The sun’s total and spectral irradiance for solar energy applications and solar radiation models. Sol. Energy 2004, 76, 423–453. [Google Scholar]
- Peter, L.M. Characterization and modeling of dye-sensitized solar cells. J. Phys. Chem. C 2007, 111, 6601–6612. [Google Scholar]
- Matthews, D.; Infelta, P.; Gratzel, M. Calculation of the photocurrent-potential characteristic for regenerative, sensitized semiconductor electrodes. Sol. Energy Mater. Sol. Cells 1996, 44, 119–155. [Google Scholar]
- Anderson, N.A.; Lian, T. Ultrafast electron injection from metal polypyridyl complexes to metal-oxide nanocrystalline thin films. Coord. Chem. Rev. 2004, 248, 1231–1246. [Google Scholar]
- Koops, S.E.; O’Regan, B.C.; Barnes, P.R.F.; Durrant, J.R. Parameters influencing the efficiency of electron injection in dye-sensitized solar cells. J. Am. Chem. Soc. 2009, 131, 4808–4818. [Google Scholar]
- Chen, R.; Yang, X.; Tian, H.; Wang, X.; Hagfeldt, A.; Sun, L. Effect of tetrahydroquinoline dyes structure on the performance of organic dye-sensitized solar cells. Chem. Mater. 2007, 19, 4007–4015. [Google Scholar]
- Oprea, C.I.; Dumbrava, A.; Enache, I.; Lungu, J.; Georgescu, A.; Moscalu, F.; Oprea, C.; Gîrţu, M.A. Role of energy level alignment in solar cells sensitized with a metalfree organic dye: A combined experimental and theoretical approach. Phys. Status Solidi 2011, A 208, 2467–2477. [Google Scholar]
- Oprea, C.I.; Panait, P.; Lungu, J.; Stamate, D.; Dumbrava, A.; Cimpoesu, F.; Gîrţu, M.A. DFT study of binding and electron transfer from a metal-free dye with carboxyl, hydroxyl and sulfonic anchors to a titanium dioxide nanocluster. Int. J. Photoenergy 2013, 893850:1–893850:15. [Google Scholar]
- Lazzeri, M.; Vittadini, A.; Selloni, A. Structure and energetics of stoichiometric TiO2 anatase surfaces. Phys. Rev. B 2001, 63, 155409:1–155409:9. [Google Scholar]
- Selloni, A. Crystal growth—Anatase shows its reactive side. Nat. Mater. 2008, 7, 613–615. [Google Scholar]
- Horn, M.; Schwerdtfeger, C.F.; Meagher, E.P. Refinement of the structure of anatase at several temperatures. Z. Krist. 1972, 136, 273–281. [Google Scholar]
- Lazzeri, M.; Selloni, A. Stress-driven reconstruction of an oxide surface: The anatase TiO2(001)-(1 × 4) surface. Phys. Rev. Lett. 2001, 87, 266105:1–266105:4. [Google Scholar]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Nazeeruddin, M.K.; Gratzel, M. First-principles modeling of the adsorption geometry and electronic structure of Ru(II) dyes on extended TiO2 substrates for dye-sensitized solar cell applications. J. Phys. Chem. C 2010, 114, 6054–6061. [Google Scholar]
- Pastore, M.; de Angelis, F. Computational modelling of TiO2 surfaces sensitized by organic dyes with different anchoring groups: Adsorption modes, structure and implication for electron injection/recombination. Phys. Chem. Chem. Phys. 2012, 14, 920–928. [Google Scholar]
- Vittadini, A.; Selloni, A.; Rotzinger, F.P.; Gratzel, M. formic acid adsorption on dry and hydrated TiO2 anatase (101) surfaces by DFT calculations. J. Phys. Chem. B 2000, 104, 1300–1306. [Google Scholar]
- Srinivas, K.; Yesudas, K.; Bhanuprakash, K.; Rao, V.J.; Giribabu, L. A combined experimental and computational investigation of anthracene based sensitizers for DSSC: Comparison of cyanoacrylic and malonic acid electron withdrawing groups binding onto the TiO2 anatase (101) surface. J. Phys. Chem. C 2009, 113, 20117–20126. [Google Scholar]
- Leon, C.P.; Kador, L.; Peng, B.; Thelakkat, M. Characterization of the adsorption of Ru-bpy dyes on mesoporous TiO2 films with UV-Vis, Raman, and FTIR spectroscopies. J. Phys. Chem. B 2006, 110, 8723–8730. [Google Scholar]
- Martsinovich, N.; Troisi, A. High-throughput computational screening of chromophores for dye-sensitized solar cells. J. Phys. Chem. C 2011, 115, 11781–11792. [Google Scholar]
- Lungu, J.; Oprea, C.I.; Dumbrava, A.; Enache, I.; Georgescu, A.; Rădulescu, C.; Ioniţă, I.; Cimpoca, G.V.; Gîrţu, M.A. Heterocyclic azodyes as pigments for dye sensitized solar cells—A combined experimental and theoretical study. J. Optoelectr. Adv. Mater. 2010, 12, 1969–1975. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oprea, C.I.; Panait, P.; Cimpoesu, F.; Ferbinteanu, M.; Gîrţu, M.A. Density Functional Theory (DFT) Study of Coumarin-based Dyes Adsorbed on TiO2 Nanoclusters—Applications to Dye-Sensitized Solar Cells. Materials 2013, 6, 2372-2392. https://doi.org/10.3390/ma6062372
Oprea CI, Panait P, Cimpoesu F, Ferbinteanu M, Gîrţu MA. Density Functional Theory (DFT) Study of Coumarin-based Dyes Adsorbed on TiO2 Nanoclusters—Applications to Dye-Sensitized Solar Cells. Materials. 2013; 6(6):2372-2392. https://doi.org/10.3390/ma6062372
Chicago/Turabian StyleOprea, Corneliu I., Petre Panait, Fanica Cimpoesu, Marilena Ferbinteanu, and Mihai A. Gîrţu. 2013. "Density Functional Theory (DFT) Study of Coumarin-based Dyes Adsorbed on TiO2 Nanoclusters—Applications to Dye-Sensitized Solar Cells" Materials 6, no. 6: 2372-2392. https://doi.org/10.3390/ma6062372
APA StyleOprea, C. I., Panait, P., Cimpoesu, F., Ferbinteanu, M., & Gîrţu, M. A. (2013). Density Functional Theory (DFT) Study of Coumarin-based Dyes Adsorbed on TiO2 Nanoclusters—Applications to Dye-Sensitized Solar Cells. Materials, 6(6), 2372-2392. https://doi.org/10.3390/ma6062372