
A Comparative Study of the Sintering Behavior of Pure and Manganese-Substituted Hydroxyapatite

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Powder Characterization

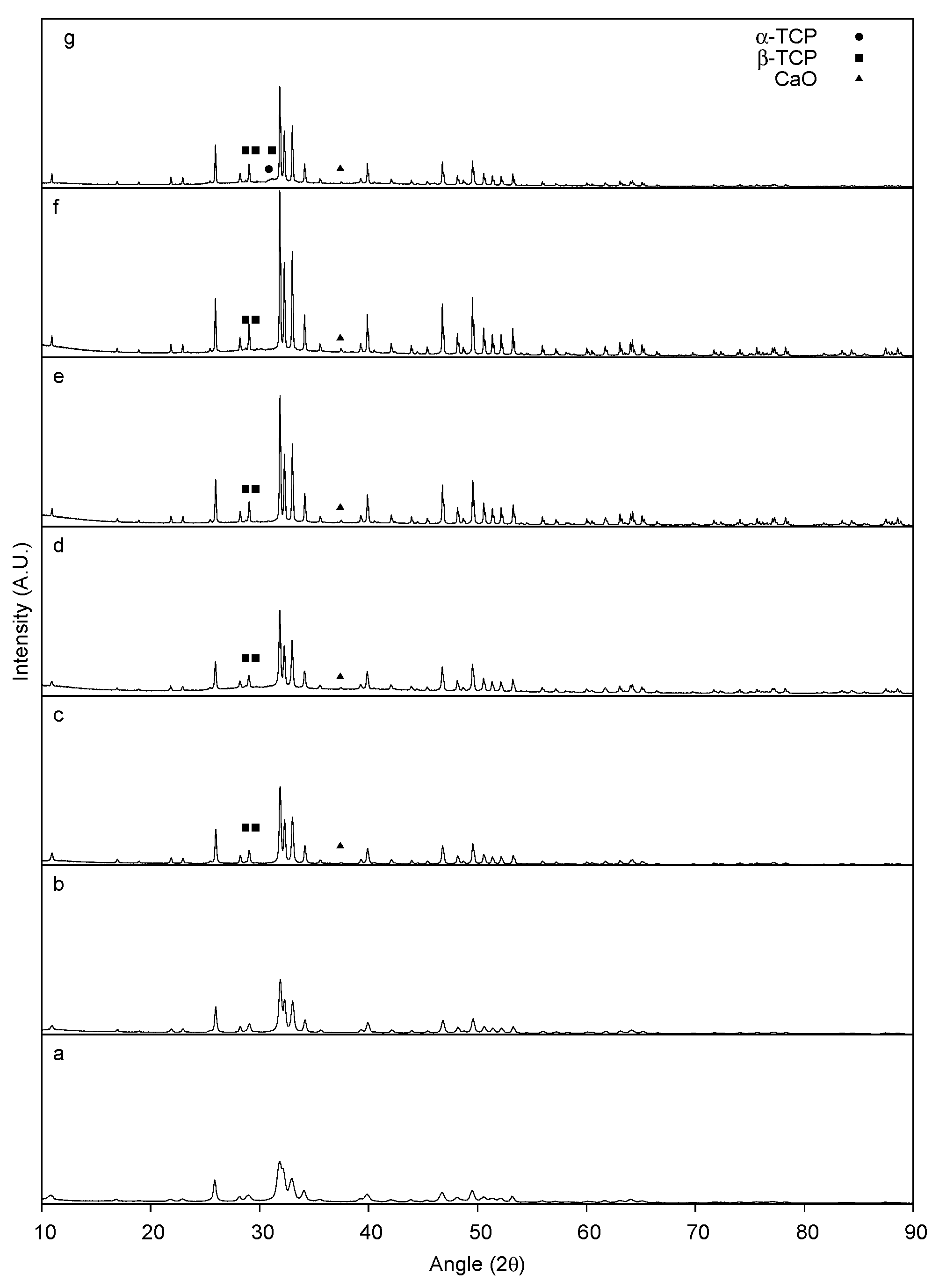
2.1.2. XRD Characterization

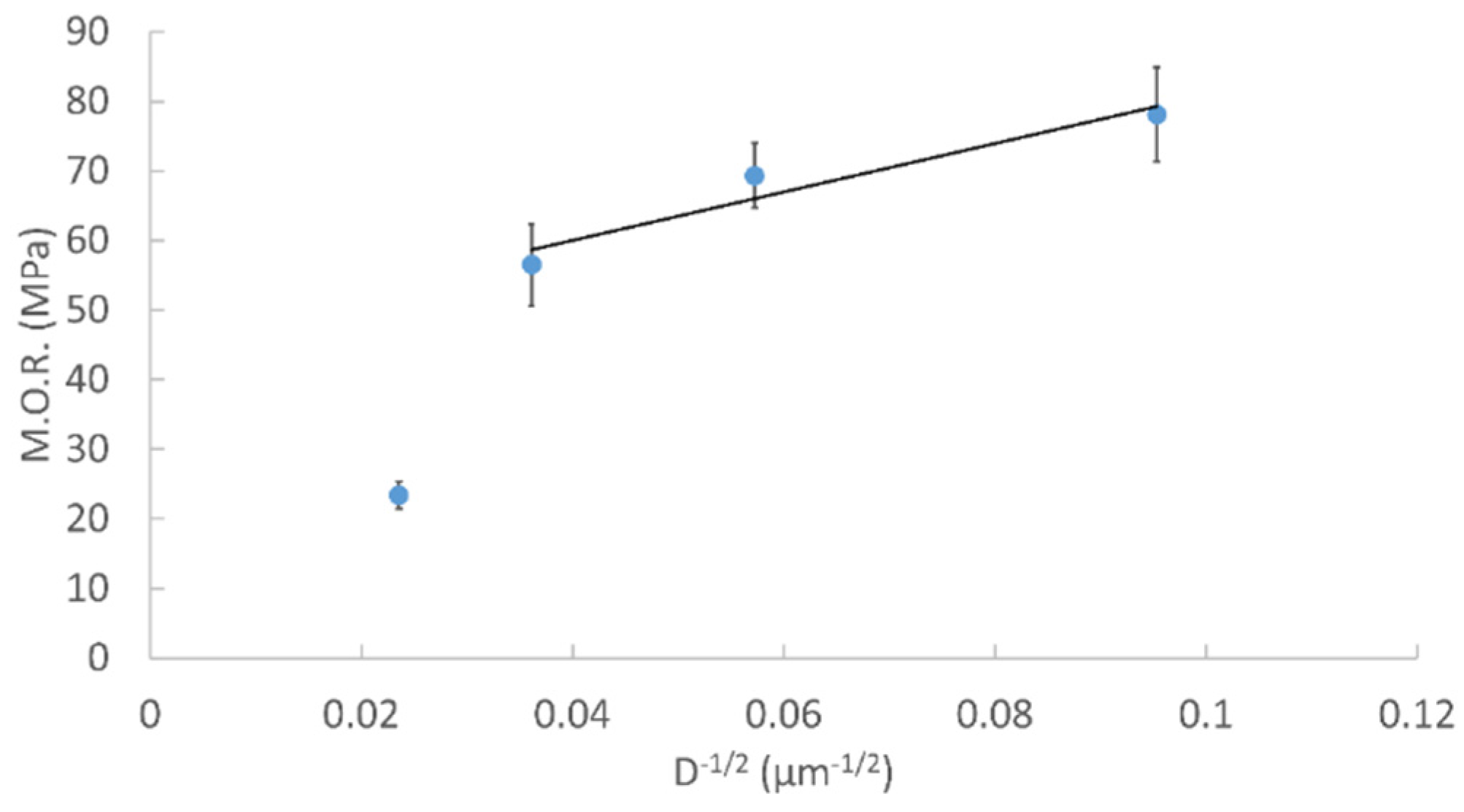
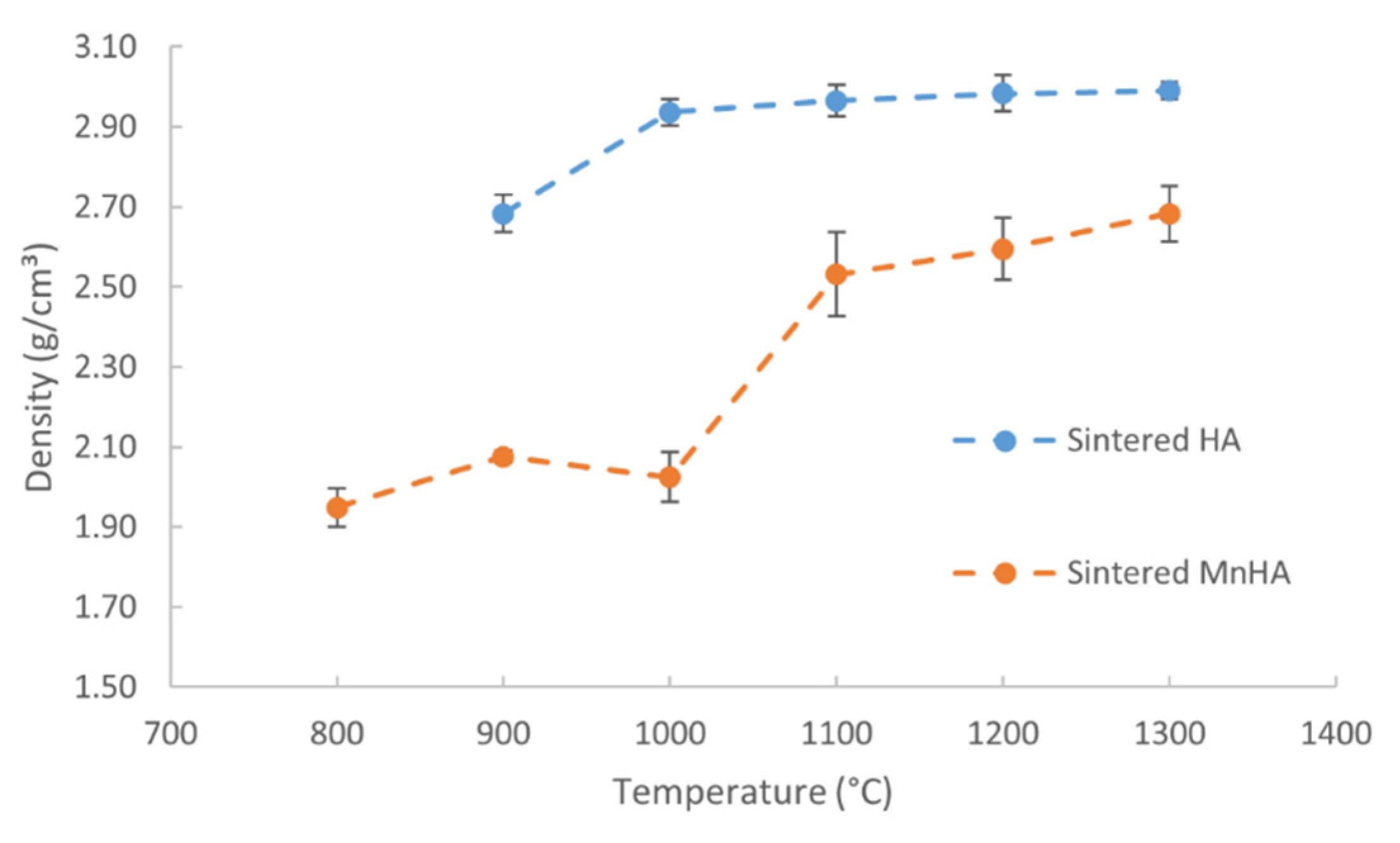
2.1.3. Density and Biaxial Flexural Strength



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | Average Green Density (g/cm3) | Average Sintered Density (g/cm3) | Average Percent Densified | |||
|---|---|---|---|---|---|---|
| HA | MnHA | HA | MnHA | HA | MnHA | |
| 800 | – | 1.78 | – | 1.95 | – | 9.4 |
| 900 | 1.80 | 1.76 | 2.68 | 2.08 | 49.4 | 18.2 |
| 1000 | 1.81 | 1.58 | 2.94 | 2.02 | 62.0 | 28.1 |
| 1100 | 1.83 | 1.62 | 2.97 | 2.53 | 61.9 | 56.4 |
| 1200 | 1.80 | 1.59 | 2.98 | 2.59 | 65.4 | 62.9 |
| 1300 | 1.81 | 1.58 | 2.99 | 2.68 | 65.5 | 70.2 |

2.1.4. FESEM Characterization




2.2. Discussion
3. Experimental Section
3.1. Materials
3.2. Hydroxyapatite and Manganese Hydroxyapatite Synthesis
3.3. Pellet Preparation and Sintering
3.4. Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Silverthorn, D.U. Human Physiology: An Integrated Approach, 5th ed.; Pearson Benjamin Cummings: San Francisco, CA, USA, 2010. [Google Scholar]
- Bose, S.; Roy, M.; Bandyopadhyay, A. Recent advances in bone tissue engineering scaffolds. Trends Biotechnol. 2012, 30, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Murugan, R.; Ramakrishna, S. Bioresorbable composite bone paste using polysaccharide based nano hydroxyapatite. Biomaterials 2004, 25, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, J.; Gremillard, L. Ceramics for medical applications: A picture for the next 20 years. J. Eur. Ceram. Soc. 2009, 29, 1245–1255. [Google Scholar] [CrossRef]
- Woodard, J.R.; Hilldore, A.J.; Lan, S.K.; Park, C.J.; Morgan, A.W.; Eurell, J.A.C.; Clark, S.G.; Wheeler, M.B.; Jamison, R.D.; Johnson, A.J.W. The mechanical properties and osteoconductivity of hydroxyapatite bone scaffolds with multi-scale porosity. Biomaterials 2007, 28, 45–54. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Standard, O.C.; Huang, T.Y.; Latella, B.A.; Swain, M.V. Mechanical behaviour of porous hydroxyapatite. Acta Biomater. 2008, 4, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Ruys, A.J.; Wei, M.; Sorrell, C.G.; Dickson, M.R.; Brandwood, A.; Milthorpe, B.K. Sintering effects on the strength of hydroxyapatite. Biomaterials 1995, 6, 409–415. [Google Scholar] [CrossRef]
- Kaur, G.; Pandey, O.P.; Singh, K.; Homa, D.; Scott, B.; Pickrell, G. A review of bioactive glasses: Their structure, properties, fabrication, and apatite formation. J. Biomed. Mater. Res. A 2014, 102, 254–274. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, N.Y. Characterisation, thermal stability and sintering of hydroxyapatite powders prepared by different routes. Mater. Chem. Phys. 2005, 94, 333–341. [Google Scholar] [CrossRef]
- Guo, L.; Huang, M.; Zhang, X. Effects of sintering temperature on structure of hydroxyapatite studied with Rietveld method. J. Mater. Sci. Mater. Med. 2003, 14, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Vladescu, A.; Birlik, I.; Braic, V.; Toparli, M.; Celik, E.; Ak Azem, F. Enhancement of the mechanical properties of hydroxyapatite by SiC addition. J. Mech. Behav. Biomed. Mater. 2014, 40, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.; Jacobsohn, O.; Niazov, T.; Werckmann, J.; Iliescu, M.; Richard-Plouet, M.; Burghaus, O.; Reinen, D. Manganese in Precipitated Hydroxyapatite. Eur. J. Inorg. Chem. 2003, 7, 1445–1451. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Capuccini, C.; Gazzano, M. Strontium-substituted hydroxyapatite nanocrystals. Inorg. Chem. Acta 2007, 360, 1009–1016. [Google Scholar] [CrossRef]
- Medvecky, L.; Stulajterova, R.; Parilak, L.; Trpcevska, J.; Durisin, J.; Barinov, S.M. Influence of manganese on stability and particle growth of hydroxyapatite in simulated body fluid. Colloids Surf. A Physicochem. Eng. Asp. 2006, 291, 221–229. [Google Scholar] [CrossRef]
- Aminzare, M.; Eskandari, A.; Baroonian, M.H.; Berenov, A.; Razavi Hesabi, Z.; Taheri, M.; Sadrnezhaad, S.K. Hydroxyapatite nanocomposites: Synthesis, sintering and mechanical properties. Ceram. Int. 2013, 39, 2197–2206. [Google Scholar] [CrossRef]
- Oshida, Y. Hydroxyapatite: Synthesis and Applications; Momentum Press: New York, NY, USA, 2015. [Google Scholar]
- Tarafder, S.; Balla, V.K.; Davies, N.M.; Bandyopadhyay, A.; Bose, S. Microwave-sintered 3D printed tricalcium phosphate scaffolds for bone tissue engineering. J. Tissue Eng. Regen. Med. 2013, 7, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Leventouri, T. Synthetic and biological hydroxyapatites: Crystal structure questions. Biomaterials 2006, 27, 3339–3342. [Google Scholar] [CrossRef] [PubMed]
- Place, E.S.; Evans, N.D.; Stevens, M.M. Complexity in biomaterials for tissue engineering. Nat. Mater. 2009, 8, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Afshari, F.T.; Kwok, J.C.; Andrews, M.R.; Faissner, A.; Ffrench-Constant, C.; Fawcett, J.W. Integrin activation or alpha 9 expression allows retinal pigmented epithelial cell adhesion on Bruch’s membrane in wet age-related macular degeneration. Brain 2010, 133, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; DeStefano, M.J. Manganese stimulates adhesion and spreading of mouse sarcoma I ascites cells. J. Cell Biol. 1973, 59, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gu, B.; Zhu, W.; Zhu, L. Integrin mediated osteoblastic adhesion on porous manganese-incorporated TiO2 coating prepared by plasma electrolytic oxidation. Exp. Ther. Med. 2013, 6, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, W.; Hamada, Y.; Kawaguchi, N.; Mori, S.; Daito, K.; Uchinaka, A.; Matsumoto, T.; Kajima, Y.; Daito, M.; Nakano, T.; et al. Synthesis of hydroxyapatite containing manganese and its evaluation of biocompatibility. Nano Biomed. 2010, 2, 37–46. [Google Scholar]
- Bigi, A.; Bracci, B.; Cuisiniere, F.; Elkaim, R.; Fini, M.; Mayer, I.; Mihailescu, I.N.; Socol, G.; Sturba, L.; Torricelli, P. Human osteoblast response to pulsed laser deposited calcium phosphate coatings. Biomaterials 2005, 26, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Erikson, K.M.; Syversen, T.; Aschner, J.L.; Aschner, M. Interactions between excessive manganese exposures and dietary iron-deficiency in neurodegeneration. Environ. Toxicol. Pharmacol. 2005, 19, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Baly, D.L.; Keen, C.L.; Hurley, L.C. Effects of manganese deficiency on pyruvate carboxylase and phosphoenolpyruvate carboxykinase activity and carbohydrate homeostasis in adult rats. Biol. Trace Element Res. 1986, 11, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Svineng, G.; Wennerberg, K.; Fassler, R.; Johansson, S. Expression of integrin subunit β1B in integrin β1-deficient GD25 cells does not interfere with αVβ3 functions. Exp. Cell Res. 2000, 254, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bracci, B.; Torricelli, P.; Panzavolta, S.; Boanini, E.; Giardino, R.; Bigi, A. Effect of Mg2+, Sr2+, and Mn2+ on the chemico-physical and in vitro biological properties of calcium phosphate biomimetic coatings. J. Inorg. Biochem. 2009, 103, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Golden, D.C.; Ming, D.W. Nutrient substituted hydroxyapatites: Synthesis and characterization. Soil Sci. Soc. Am. J. 1999, 63, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Sutter, B.; Wasowicz, T.; Howard, T.; Hossner, L.R.; Ming, D.W. Characterization of iron, manganese, and copper synthetic hydroxyapatites by electron paramagnetic resonance spectroscopy. Soil Sci. Soc. Am. J. 2002, 66, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Sutter, B.; Hossner, L.R.; Ming, D.W. Dissolution Kinetics of iron-, manganese-, and copper-containing synthetic hydroxyapatites. Soil Sci. Soc. Am. J. 2005, 69, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Sutter, B.; Ming, D.W.; Clearfield, A.; Hossner, L.R. Mineralogical and chemical characterization of iron-, manganese-, and copper-containing synthetic hydroxyapatites. Soil Sci. Soc. Am. J. 2003, 67, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Sutter, B.; Taylor, R.E.; Hossner, L.R.; Ming, D.W. Solid state 31phosphorous nuclear magnetic resonance of iron-, manganese-, and copper-containing synthetic hydroxyapatites. Soil Sci. Soc. Am. J. 2002, 66, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.; Cuisinier, F.J.G.; Popov, I.; Schleich, Y.; Gdalya, S.; Burghaus, O.; Reinen, D. Phase relations between β-tricalcium phosphate and hydroxyapatite with manganese (II): Structural and spectroscopic properties. Eur. J. Inorg. Chem. 2006, 2006, 1460–1465. [Google Scholar] [CrossRef]
- Mayer, I.; Cuisinier, F.J.G.; Gdalya, S.; Popov, I. TEM study of the morphology of Mn2+ doped calcium hydroxyapatite and β-tricalcium phosphate. J. Inorg. Biochem. 2008, 102, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Paluszkiewicz, C.; Slosarczyk, A.; Pijocha, D.; Sitarz, M.; Bucko, M.; Zima, A.A.; Chroscicka, A.; Lewandowska-Szumiel, M. Synthesis, structural properties, and thermal stability of Mn-doped hydroxyapatite. J. Mol. Struct. 2010, 976, 301–309. [Google Scholar] [CrossRef]
- Li, Y.; Nam, C.T.; Ooi, C.P. Iron (III) and manganese (II) substituted hydroxyapatite nanoparticles: Characterization and cytotoxicity analysis. J. Phys. Conf. Ser. 2009, 187. [Google Scholar] [CrossRef]
- Li, Y.; Widodo, J.; Lim, S.; Ooi, C.P. Synthesis and cyctocompatability of managanese (II) and iron (III) substituted hydroxyapatite nanoparticles. J. Mater. Sci. 2012, 471, 754–763. [Google Scholar] [CrossRef]
- Ramesh, S.; Tan, C.Y.; Peralta, C.L.; Teng, W.D. The effect of manganese oxide on the sinterability of hydroxyapatite. Sci. Technol. Adv. Mater. 2007, 8, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Muralithran, G.; Ramesh, S. The effects of sintering temperature on the properties of hydroxyapatite. Ceram. Int. 2000, 26, 221–230. [Google Scholar] [CrossRef]
- Barinov, S.; Rau, J.; Nunziante Cesaro, S.; Durisin, J.; Fadeeva, I.; Ferro, D.; Medvecky, L.; Trionfetti, G. Carbonate release from carbonated hydroxyapatite in the wide temperature rage. J. Mater. Sci. Mater. Med. 2006, 17, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Zilm, M.E.; Staruch, M.; Jain, M.; Wei, M. An intrinsically magnetic biomaterial with tunable magnetic properties. J. Mater. Chem. B 2014, 2, 7176–7185. [Google Scholar] [CrossRef]
- Gillot, B.; El Guendouzi, M.; Laarj, M. Particle size effects on the oxidation-reduction behavior of Mn3O4 hausmannite. Mater. Chem. Phys. 2001, 70, 54–60. [Google Scholar] [CrossRef]
- Carrodeguass, R.G.; de Aza, S. α-Tricalcium phosphate: Synthesis, properties and biomedical applications. Acta Biomater. 2011, 7, 3536–3546. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shaw, L. Morphology-enhanced low temperature sintering of nanocrystalline hydroxyapatite. Adv. Mater. 2007, 19, 2364–2369. [Google Scholar] [CrossRef]
- Pattanayak, D.K.; Divya, P.; Upadyay, S.; Prasad, R.C.; Rao, B.T.; Rama Mohan, T.R. Synthesis and evaluation of hydroxyapatite ceramics. Trends Biomater. Artif. Organs 2005, 18, 87–92. [Google Scholar]
- Pattanayak, D.K.; Dash, R.; Prasad, R.C.; Rao, B.T.; Rama Mahan, T.R. Synthesis and sintered properties evaluation of calcium phosphate ceramics. Mater. Sci. Eng. C 2007, 27, 654–690. [Google Scholar] [CrossRef]
- Akao, M.; Aoki, H.; Kato, K. Mechanical properties of sintered hydroxyapatite for prosthetic applications. J. Mater. Sci. 1981, 16, 809–812. [Google Scholar] [CrossRef]
- Pramanik, S.; Agarwal, A.K.; Rai, K.N.; Garg, A. Development of high strength hydroxyapatite by solid-state-sintering process. Ceram. Int. 2007, 33, 419–426. [Google Scholar] [CrossRef]
- Sureshbabu, S.; Komath, M.; Varma, H.K. In situ formation of hydroxyapatite—alpha tricalcium phosphate biphasic ceramic with higher strength and bioactivity. J. Am. Ceram. Soc. 2012, 95, 915–924. [Google Scholar]
- Kannan, S.; Vieira, S.I.; Olhero, S.M.; Torres, P.M.C.; da Cruz e Silva, O.A.B.; Ferreira, J.M.F.; Pina, S. Synthesis, mechanical and biological characterization of ionic doped carbonated hydroxyapatite/ß-tricalcium phosphate mixtures. Acta Biomater. 2011, 7, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.S.; Chin, T.S.; Lai, L.S.; Chiu, S.Y.; Chung, K.H.; Chang, C.S.; Lui, M.T. Hydroxyapatite synthesized by a simplified hydrothermal method. Ceram. Int. 1997, 23, 19–25. [Google Scholar] [CrossRef]
- Boilet, L.; Descamps, M.; Rguiti, E.; Tricoteaux, A.; Lu, J.; Petit, F.; Lardot, V.; Cambier, F.; Leriche, A. Processing and properties of transparent hydroxyapatite and ß-tricalcium phosphate obtained by HIP process. Ceram. Int. 2013, 39, 283–288. [Google Scholar] [CrossRef]
- Raynaud, S.; Champion, E.; Lafon, J.P.; Bernache-Assollant, D. Calcium phosphate apatites with variable Ca/P atomic ratio III. Mechanical properties and degradation in solution of hot pressed ceramics. Biomaterials 2002, 23, 1081–1089. [Google Scholar] [CrossRef]
- An, Y.H.; Draugh, R.A. Mechanical Testing of Bone and the Bone-Implant Interface; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Wang, J.; Shaw, L. Nanocrystalline Hydroxyapatite with simultaneous enhancements in hardness and toughness. Biomaterials 2009, 30, 6565–6572. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.; Zilm, M.; Wei, M. A Comparative study of the sintering behavior of pure and iron-substituted hydroxyapatite. Bioceram. Dev. Appl. 2013, 3. [Google Scholar] [CrossRef]
- Yoshida, K.; Kondo, N.; Kita, H.; Mitamura, M.; Hashimoto, K.; Toda, Y. Effect of substitutional monovalent and divalent metal ions on mechanical properties of ß-tricalcium phosphate. J. Am. Ceram. Soc. 2005, 88, 2315–2318. [Google Scholar] [CrossRef]
- Tanimoto, Y.; Nishiyama, N. Preparation and physical properties of tricalcium phosphate laminates for bone tissue engineering. J. Biomed. Mater. Res. A 2008, 85, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Seol, Y.J.; Park, J.Y.; Jung, J.W.; Jang, J.; Girdhari, R.; Kim, S.W.; Cho, D.W. Improvement of bone regeneration capability of ceramic scaffolds by accelerated release of their calcium ions. Tissue Eng. A 2014, 20, 2840–2849. [Google Scholar] [CrossRef] [PubMed]
- ASTM Standard F 394 Test Method for Biaxial Flexure Strength (Modulus of Rupture) of Ceramic Substrates; ASTM International: West Conshohocken, PA, USA, 1996.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zilm, M.; Thomson, S.D.; Wei, M. A Comparative Study of the Sintering Behavior of Pure and Manganese-Substituted Hydroxyapatite. Materials 2015, 8, 6419-6436. https://doi.org/10.3390/ma8095308
Zilm M, Thomson SD, Wei M. A Comparative Study of the Sintering Behavior of Pure and Manganese-Substituted Hydroxyapatite. Materials. 2015; 8(9):6419-6436. https://doi.org/10.3390/ma8095308
Chicago/Turabian StyleZilm, Michael, Seamus D. Thomson, and Mei Wei. 2015. "A Comparative Study of the Sintering Behavior of Pure and Manganese-Substituted Hydroxyapatite" Materials 8, no. 9: 6419-6436. https://doi.org/10.3390/ma8095308