
Molecular Mobility in Hyperbranched Polymers and Their Interaction with an Epoxy Matrix


Abstract
:1. Introduction
2. Results
2.1. Optical Microscopy
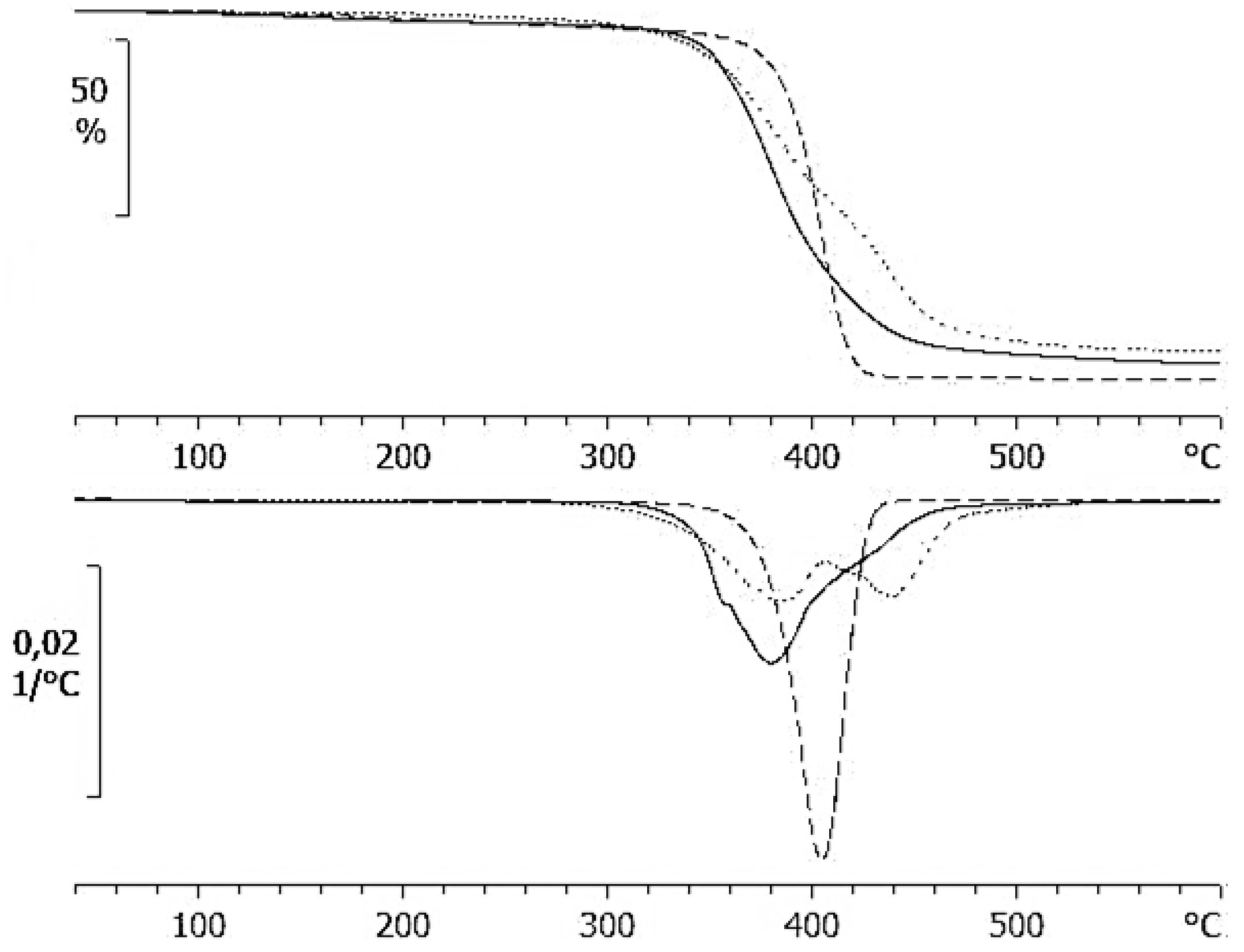
2.2. Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) Results
2.3. Dielectric Relaxation Spectroscopy (DRS) Results
2.3.1. Hyperbranched Polyethyleneimine (HBPEI)
2.3.2. Epoxy
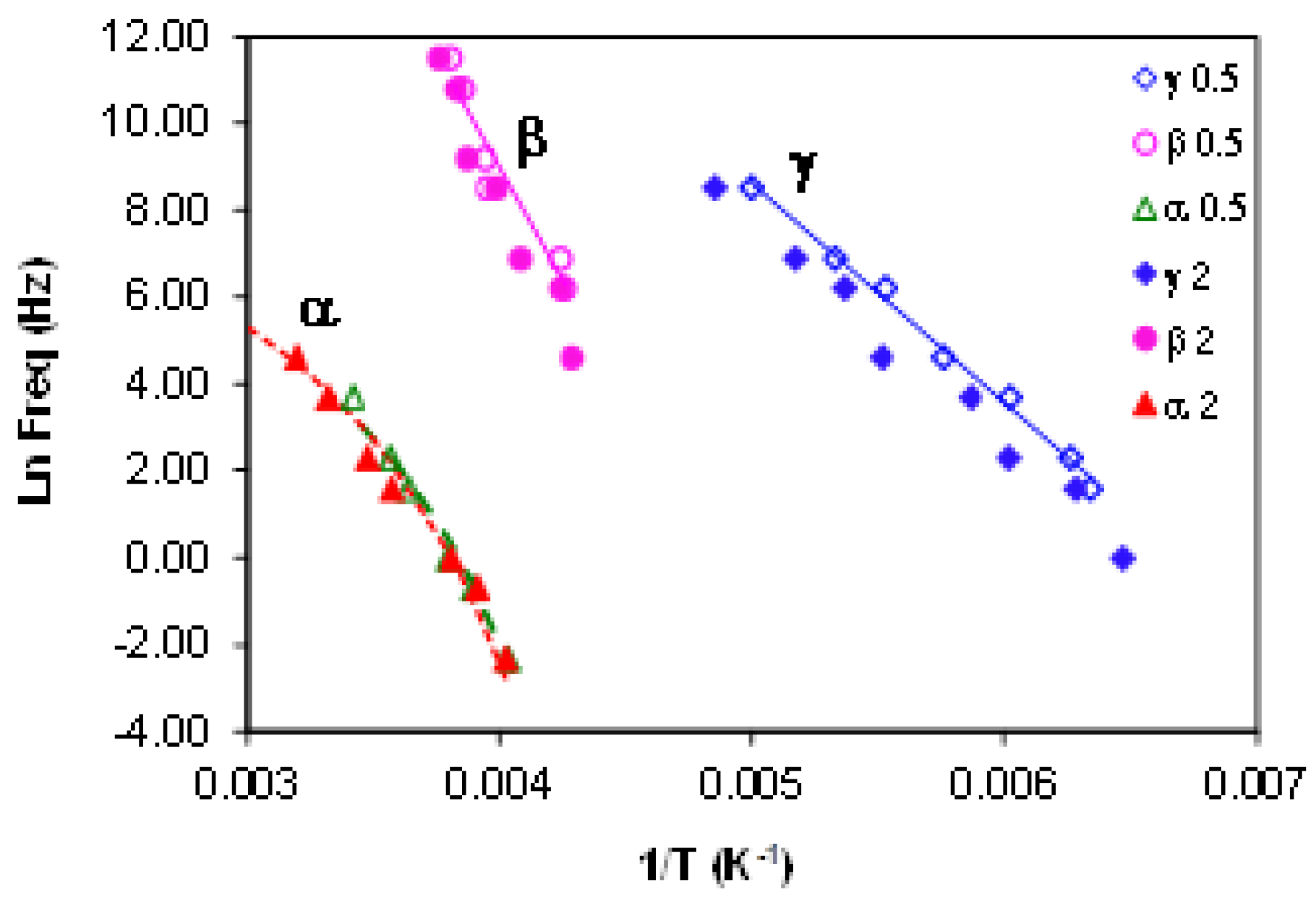
2.3.3. ELP System
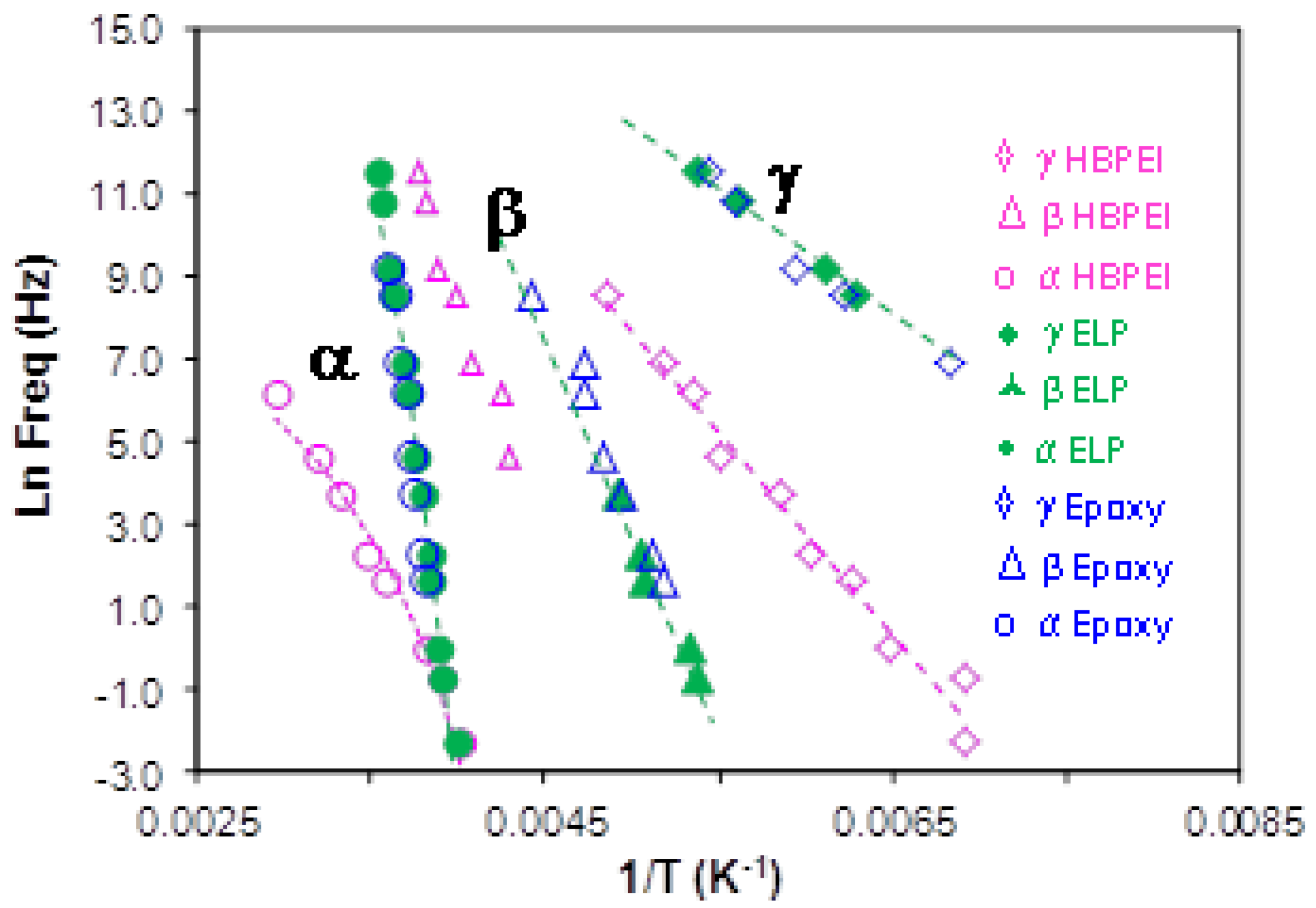
2.3.4. Comparison of HBPEI, Epoxy and Uncured ELP
2.4. Dynamic Mechanical Analysis (DMA) Results
2.4.1. Uncured Systems
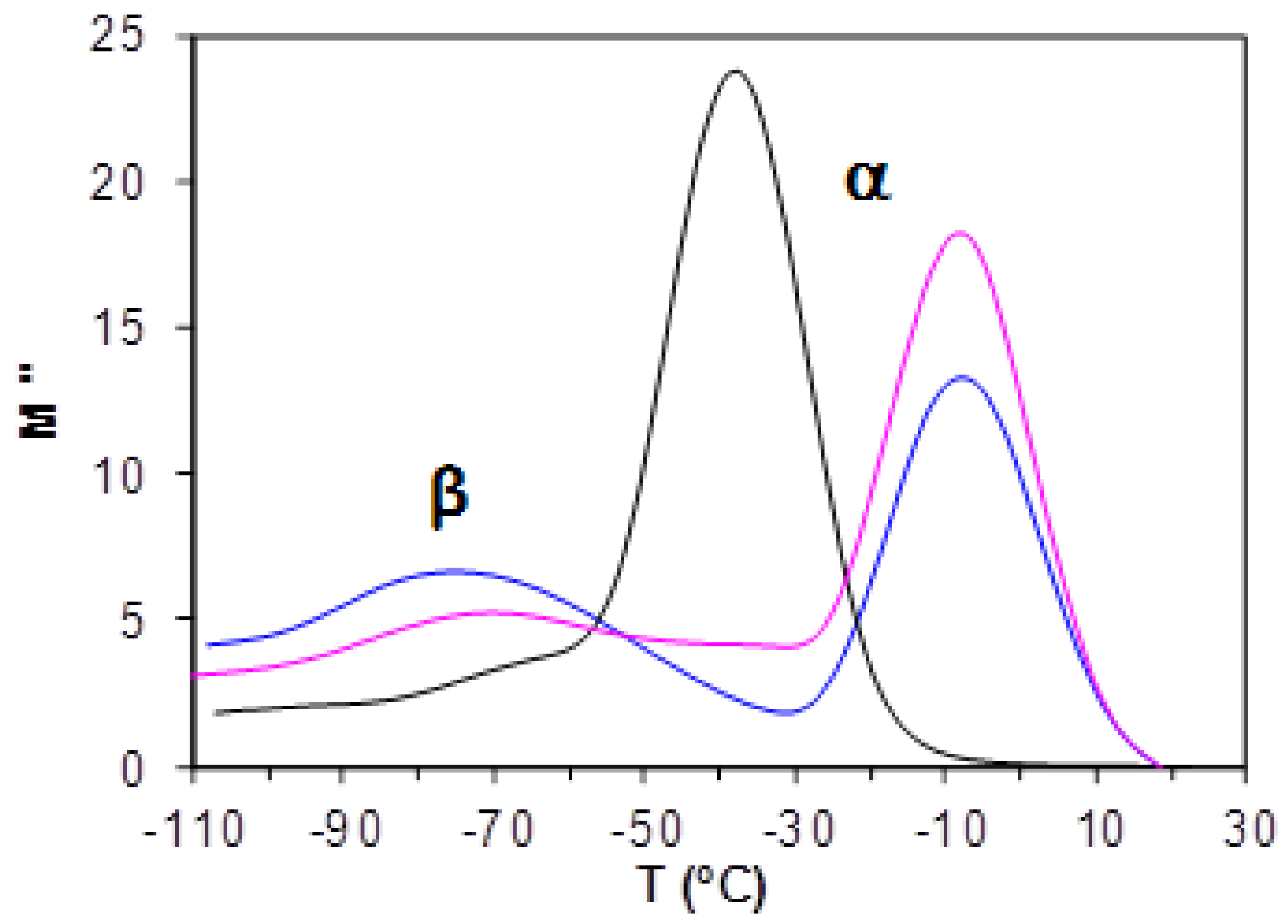
2.4.2. Cured ELP System
3. Materials and Methods
3.1. Materials
3.2. Preparation of the Samples
3.3. Experimental Techniques
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pascault, J.P.; Sautereau, H.; Verdu, J.; Williams, R.J.J. Thermosetting Polymers; Marcel Dekker: New York, NY, USA, 2002. [Google Scholar]
- Epoxy Resins: Chemistry and Technology; May, C.A. (Ed.) Marcel Dekker: New York, NY, USA, 1973.
- Thermal Characterization of Polymeric Materials; Turi, E.A. (Ed.) Academic Press: San Diego, CA, USA, 1997.
- Fernández, R.; De Arlasa, B.F.; Oyanguren, P.A.; Mondragón, I. Kinetic studies of the polymerization of an epoxy resin modified with rhodamine B. Thermochim. Acta 2009, 493, 6–13. [Google Scholar] [CrossRef]
- Bosman, A.W.; Janssen, H.M.; Meijer, E.W. About dendrimers: Structure, physical properties, and applications. Chem. Rev. 1999, 99, 1665–1688. [Google Scholar] [CrossRef] [PubMed]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A new class of polymers—Starbust-dendritic macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef]
- Fréchet, J.M.J.; Tomalia, D.A. Dendrimers and Other Dendritic Polymers; John Wiley & Sons Ltd.: New York, NY, USA, 2001. [Google Scholar]
- Tande, B.M.; Wagner, N.J.; Kim, Y.H. Poly(propylene imine) dendrimers as plasticizers for polyvinyl chloride. J. Polym. Sci. B Polym. Phys. 2007, 45, 1970–1975. [Google Scholar] [CrossRef]
- Okazaki, M.; Murota, M.; Kawaguchi, Y.; Tsubokawa, N. Curing of epoxy resin by ultrafine silica modified by grafting of hyperbranched polyamidoamine using dendrimer synthesis methodology. J. Appl. Polym. Sci. 2001, 80, 573–579. [Google Scholar] [CrossRef]
- Ukaji, M.; Takamura, M.; Shirai, K.; Gang, W.; Yamauchi, T.; Tsubokawa, N. Curing of epoxy resin by hyperbranched poly(amidoamine)grafted silica nanoparticles and their properties. Polym. J. 2008, 40, 607–613. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, D.; Fu, R.; He, P. Behavior of polyamidoamine dendrimers as curing agents in bis-phenol a epoxy resin systems. Polym. Int. 2005, 54, 495–499. [Google Scholar] [CrossRef]
- Cheng, Y.; Xu, T.; He, P. Polyamidoamine dendrimers as curing agents: The optimum polyamidoamine concentration selected by dynamic torsional vibration method and thermogravimetric analyses. J. Appl. Polym. Sci. 2007, 103, 1430–1434. [Google Scholar] [CrossRef]
- Xu, D.M.; Zhang, K.D.; Zhu, X.L. Curing of DGEBA epoxy resin by low generation amino-group-terminated dendrimers. J. Appl. Polym. Sci. 2006, 101, 3902–3906. [Google Scholar] [CrossRef]
- Wan, J.; Bu, Z.; Xu, C.; Li, B.; Fan, H. Learning about novel amine-adduct curing agents for epoxy resins: Butyl-glycidylether-modified poly(propyleneimine) dendrimers. Thermochim. Acta 2011, 519, 72–82. [Google Scholar] [CrossRef]
- Wan, J.T.; Fan, H.; Li, B.G.; Xu, C.J.; Bu, Z.Y. Synthesis and nonisothermal reaction of a novel acrylonitrile-capped poly(propyleneimine) dendrimer with epoxy resin. J. Therm. Anal. Calorim. 2011, 103, 685–692. [Google Scholar] [CrossRef]
- Zhou, D.P.; Wang, H.M.; Liu, D.; Wang, S.S. Isothermal Cure of Bisphenol Epoxy Resin with a Nonlinear Aliphatic Polyamine Hardener. Polym. Plast. Technol. Eng. 2012, 51, 883–888. [Google Scholar] [CrossRef]
- Voit, B. New developments in hyperbranched polymers. J. Polym. Sci. A Polym. Chem. 2000, 38, 2505–2525. [Google Scholar] [CrossRef]
- Yates, C.R.; Hayes, W. Synthesis and applications of hyperbranched polymers. Eur. Polym. J. 2004, 40, 1257–1281. [Google Scholar] [CrossRef]
- Oh, J.H.; Jang, J.S.; Lee, S.H. Curing behavior of tetrafunctional epoxy resin/hyperbranched polymer system. Polymer 2001, 42, 8339–8347. [Google Scholar] [CrossRef]
- Foix, D.; Yu, Y.; Serra, A.; Ramis, X.; Salla, J.M. Study on the chemical modification of epoxy/anhydride thermosets using a hydroxyl terminated hyperbranched polymer. Eur. Polym. J. 2009, 45, 1454–1466. [Google Scholar] [CrossRef]
- Fernandez-Francos, X.; Salla, J.M.; Cadenato, A.; Morancho, J.M.; Serra, A.; Mantecón, A.; Ramis, X. A new strategy for controlling shrinkage of DGEBA resins cured by cationic copolymerization with hydroxyl-terminated hyperbranched polymers and ytterbium triflate as an initiator. J. Appl. Polym. Sci. 2009, 111, 2822–2829. [Google Scholar] [CrossRef]
- Sangermano, M.; Priola, A.; Malucelli, G.; Bongiovanni, R.; Quaglia, A.; Voit, B.; Ziemer, A. Phenolic hyperbranched polymers as additives in cationic photopolymerization of epoxy systems. Macromol. Mater. Eng. 2004, 289, 442–446. [Google Scholar] [CrossRef]
- Sangermano, M.; Malucelli, G.; Bongiovanni, R.; Priola, A.; Harden, A. Investigation on the effect of the presence of hyperbranched polymers on thermal and mechanical properties of an epoxy UV-cured system. Polym. Int. 2005, 54, 917–921. [Google Scholar] [CrossRef]
- Santiago, D.; Fernandez-Francos, X.; Ramis, X.; Salla, J.M.; Sangermano, M. Comparative curing kinetics and thermal-mechanical properties of DGEBA thermosets cured with a hyperbranched poly(ethyleneimine) and an aliphatic triamine. Thermochim. Acta 2011, 526, 9–21. [Google Scholar] [CrossRef]
- Fernandez-Francos, X.; Santiago, D.; Ferrando, F.; Ramis, X.; Salla, J.M.; Serra, A.; Sangermano, M. Network structure and thermomechanical properties of hybrid DGEBA networks cured with 1-methylimidazole and hyperbranched poly(ethyleneimine)s. J. Polym. Sci. B Polym. Phys. 2012, 50, 1489–1503. [Google Scholar] [CrossRef]
- Cortés, P.; Fraga, I.; Calventus, Y.; Román, F.; Hutchinson, J.M.; Ferrando, F. A new epoxy-based layered silicate nanocomposite using a hyperbranched polymer: Study of the curing reaction and nanostructure development. Materials 2014, 7, 1830–1849. [Google Scholar] [CrossRef] [Green Version]
- Casalini, R.; Fioretto, D.; Livi, A.; Lucchesi, M.; Rolla, P.A. Influence of the glass transition on the secondary relaxation of an epoxy resin. Phys. Rev. B 1997, 56, 3016–3021. [Google Scholar] [CrossRef]
- Capaccioli, S.; Corezzi, S.; Gallone, G.; Rolla, P.A.; Comez, L.; Fioretto, D. Dynamics of epoxies: A full dielectric analysis by wideband spectroscopy. J. Non-Cryst. Sol. 1998, 235–237, 576–579. [Google Scholar] [CrossRef]
- Corezzi, S.; Beiner, M.; Huth, H.; Schröter, K.; Capaccioli, S.; Casalini, R.; Fioretto, D.; Donth, E. Two crossover regions in the dynamics of glass forming epoxy resins. J. Chem. Phys. 2002, 117, 2435–2448. [Google Scholar] [CrossRef]
- Beiner, M.; Ngai, K.L. Interrelation between primary and secondary relaxations in polymerizing systems based on epoxy resins. Macromolecules 2005, 38, 7033–7042. [Google Scholar] [CrossRef]
- Román, F.; Calventus, Y.; Colomer, P.; Hutchinson, J.M. Dielectric relaxation spectroscopy of epoxy resin cured with a hyperbranched poly(ethyleneimine). In Book of Abstracts, Proceedings of the 2nd Central and Eastern European Conference on Thermal Analysis and Calorimetry, Vilnius, Lithuania, 27–30 August 2013; p. 169.
- Gallone, G.; Capaccioli, S.; Levita, G.; Rolla, P.A.; Corezzi, S. Dielectric analysis of the linear polymerization of an epoxy resin. Polym. Int. 2001, 50, 545–551. [Google Scholar] [CrossRef]
- Senturia, S.D.; Sheppard, N.F. Dielectric analysis of thermoset cure. Adv. Polym. Sci. 1986, 80, 1–47. [Google Scholar]
- Mangion, M.B.M.; Johari, G.P. Relaxations of thermosets. 3. Sub-Tg dielectric relaxations of bisphenol-A based epoxide cured with different cross-linking agents. J. Polym. Sci. B Polym. Phys. 1990, 28, 71–83. [Google Scholar] [CrossRef]
- Mangion, M.B.M.; Johari, G.P. Relaxations of thermosets. 4. A dielectric study of cross-linking of diglycidyl ether of bisphenol-A by 2 curing agents. J. Polym. Sci. B Polym. Phys. 1990, 28, 1621–1639. [Google Scholar] [CrossRef]
- Mangion, M.B.M.; Johari, G.P. Relaxations of thermosets. 6. Effects of cross-linking on sub-Tg relaxations during the curing and aging of epoxide-based thermosets. J. Polym. Sci. B Polym. Phys. 1991, 29, 437–449. [Google Scholar] [CrossRef]
- Koike, T. Dielectric relaxation during isothermal curing of epoxy resin with an aromatic amine. J. Appl. Polym. Sci. 1992, 44, 679–690. [Google Scholar] [CrossRef]
- Parthun, M.G.; Johari, G.P. Relaxations in thermosets. 23. Dielectric studies of curing kinetics of an epoxide with diamines of varying chain lengths. Macromolecules 1992, 25, 3254–3265. [Google Scholar] [CrossRef]
- Fournier, J.; Williams, G.; Duch, C.; Aldridge, G.A. Changes in molecular dynamics during bulk polymerization of an epoxide-amine system as studied by dielectric relaxation spectroscopy. Macromolecules 1996, 29, 7097–7107. [Google Scholar] [CrossRef]
- Wasylyshyn, D.A.; Johari, G.P. Physical aspects of network polymerization from calorimetry and dielectric spectroscopy of a triepoxide reacting with different monoamines. J. Polym. Sci. B Polym. Phys. 1997, 35, 437–456. [Google Scholar] [CrossRef]
- Tombari, E.; Ferrari, C.; Salvetti, G.; Johari, G.P. Thermodynamics and dielectric relaxation during the polymerization of a flexible diamine-diepoxide mixture and its glassy-state relaxation. Phys. Chem. Chem. Phys. 1999, 1, 1965–1970. [Google Scholar] [CrossRef]
- Deng, Y.; Martin, G.C. Modeling diffusion during thermoset cure—An approach based on dielectric analysis. Macromolecules 1994, 27, 5141–5146. [Google Scholar] [CrossRef]
- Parthun, M.G.; Johari, G.P. Dynamics of a molecules growth—Ultrasonic relaxation studies. J. Chem. Phys. 1995, 102, 6301–6307. [Google Scholar] [CrossRef]
- Parthun, M.G.; Johari, G.P. Dielectric spectroscopy of a polymerizing liquid and the evolution of molecular dynamics with increase in the number of covalent bonds. J. Chem. Phys. 1995, 103, 440–450. [Google Scholar] [CrossRef]
- Wasylyshyn, D.A.; Johari, G.P. The effects of covalent bonds on the localized relaxations in the glassy states of linear chain and network macromolecules. J. Chem. Phys. 1996, 104, 5683–5689. [Google Scholar] [CrossRef]
- Montserrat, S.; Román, F.; Colomer, P. Vitrification and dielectric relaxation during the isothermal curing of an epoxy-amine resin. Polymer 2003, 44, 101–114. [Google Scholar] [CrossRef]
- Fraga, I.; Montserrat, S.; Hutchinson, J.M. Vitrification during the isothermal cure of thermosets: Comparison of theoretical simulations with temperature-modulated DSC and dielectric analysis. Macromol. Chem. Phys. 2008, 209, 2003–2011. [Google Scholar] [CrossRef]
- Zong, L.; Zhou, S.; Sgriccia, N.; Hawley, M.C.; Sun, R.; Kempel, L.C. Dielectric properties of an epoxy-amine system at a high microwave frequency. Polym. Eng. Sci. 2005, 45, 1576–1580. [Google Scholar] [CrossRef]
- Shimbo, M.; Ochi, M.; Iesako, H. Mechanical relaxation mechanism of epoxide resins cured with acid anhydrides. J. Polym. Sci. B Polym. Phys. 1984, 22, 1461–1470. [Google Scholar] [CrossRef]
- Ochi, M.; Yoshizumi, M.; Shimbo, M. Mechanical and dielectric relaxations of epoxide resins containing the spiro-ring structure. 2. Effect of the introduction of methoxy branches on low temperature relaxations. J. Polym. Sci. B Polym. Phys. 1987, 25, 1817–1827. [Google Scholar] [CrossRef]
- Trahasch, B.; Stühn, B.; Frey, H.; Lorenz, K. Dielectric relaxation in carbosilane dendrimers with perfluorinated end groups. Macromolecules 1999, 32, 1962–1966. [Google Scholar] [CrossRef]
- Trahasch, B.; Frey, H.; Lorenz, K.; Stühn, B. Dielectric relaxation in carbosilane dendrimers with cyanobiphenyl end groups. Colloid Polym. Sci. 1999, 277, 1186–1192. [Google Scholar] [CrossRef]
- Emran, S.K.; Newkome, G.R.; Weis, C.D.; Harmon, J.P. Molecular relaxations in ester-terminated, amide-based dendrimers. J. Polym. Sci. B Polym. Phys. 1999, 37, 2025–2038. [Google Scholar] [CrossRef]
- Huwe, A.; Appelhans, D.; Prigann, J.; Voit, B.I.; Kremer, F. Broadband dielectric spectroscopy on the molecular dynamics in dendritic model systems. Macromolecules 2000, 33, 3762–3766. [Google Scholar] [CrossRef]
- Okrasa, L.; Zigon, M.; Zagar, E.; Czech, P.; Boiteux, G. Molecular dynamics of linear and hyperbranched polyurethanes and their blends. J. Non-Cryst. Sol. 2005, 351, 2753–2758. [Google Scholar] [CrossRef]
- Kyritsis, A.; Raftopoulos, K.; Abdel Rehim, M.; Shabaan, S.S.; Ghoneim, A.; Turky, G. Structure and molecular dynamics of hyperbranched polymeric systems with urethane and urea linkages. Polymer 2009, 50, 4039–4047. [Google Scholar] [CrossRef]
- Garcia-Bernabé, A.; Diaz-Calleja, R.; Haag, R. Broadband dielectric spectroscopy studies of hyperbranched polyglycerols. Macromol. Chem. Phys. 2006, 207, 970–977. [Google Scholar] [CrossRef]
- Garcia-Bernabé, A.; Dominguez-Espinosa, G.; Diaz-Calleja, R.; Riande, E.; Haag, R. Secondary and primary relaxations in hyperbranched polyglycerol: A comparative study in the frequency and time domains. J. Chem. Phys. 2007, 127, 124904:1–124904:10. [Google Scholar] [CrossRef] [PubMed]
- Malmström, E.; Liu, F.; Boyd, R.H.; Hult, A.; Gedde, U.W. Relaxation processes in hyperbranched polyesters. Polym. Bull. 1994, 32, 679–685. [Google Scholar] [CrossRef]
- Malmström, E.; Hult, A.; Gedde, U.W.; Liu, F.; Boyd, R.H. Relaxation processes in hyperbranched polyesters: Influence of terminal groups. Polymer 1997, 38, 4873–4879. [Google Scholar] [CrossRef]
- Zhu, P.W.; Zheng, S.; Simon, G. Dielectric relaxations in a hyperbranched polyester with terminal hydroxyl groups: Effects of generation number. Macromol. Chem. Phys. 2001, 202, 3008–3017. [Google Scholar] [CrossRef]
- Hakme, C.; Stevenson, I.; Fulchiron, R.; Seytre, G.; Clement, F.; Odoni, L.; Rochat, S.; Varlet, J. Dielectric studies of hyperbranched aromatic polyamide and polyamide-6,6 blends. J. Appl. Polym. Sci. 2005, 97, 1522–1537. [Google Scholar] [CrossRef]
- Sangoro, J.R.; Turky, G.; Abdel Rehim, M.; Iacob, C.; Naumov, S.; Ghoneim, A.; Karger, J.; Kremer, F. Charge transport and dipolar relaxations in hyperbranched polyamide amines. Macromolecules 2009, 42, 1648–1651. [Google Scholar] [CrossRef]
- Czech, P.; Okrasa, L.; Boiteux, G.; Mechin, F.; Ulanski, J. Polyurethane networks based on hyperbranched polyesters: Synthesis and molecular relaxations. J. Non-Cryst. Sol. 2005, 351, 2735–2741. [Google Scholar] [CrossRef]
- Czech, P.; Okrasa, L.; Mechin, F.; Boiteux, G.; Ulanski, J. Investigation of the polyurethane chain length influence on the molecular dynamics in networks crosslinked by hyperbranched polyester. Polymer 2006, 47, 7207–7215. [Google Scholar] [CrossRef]
- Okrasa, L.; Czech, P.; Boiteux, G.; Mechin, F.; Ulanski, J. Studies of molecular dynamics in polyurethane networks with hyperbranched polyester Boltorn (R) H30 as a crosslinker. J. Non-Cryst. Sol. 2007, 353, 4293–4297. [Google Scholar] [CrossRef]
- Maroulas, P.; Kripotou, S.; Sysel, P.; Hobzova, R.; Kotek, J.; Pissis, P. Molecular dynamics in hyperbranched polyimides cross-linked with ethylene glycol diglycidyl ether. J. Non-Cryst. Sol. 2005, 352, 4800–4803. [Google Scholar] [CrossRef]
- Shiravand, F.; Fraga, I.; Cortés, P.; Calventus, Y.; Hutchinson, J.M. Thermal analysis of polymer layered silicate nanocomposites Identification of nanostructure development by DSC. J. Therm. Anal. Calorim. 2014, 118, 723–729. [Google Scholar] [CrossRef]
- Grassie, N.; Guy, M.I.; Tennent, N.H. Degradation of epoxy polymers. 1. Products of thermal degradation of bisphenol-A diglycidyl ether. Polym. Degrad. Stab. 1985, 12, 65–91. [Google Scholar] [CrossRef]
- Grassie, N.; Guy, M.I.; Tennent, N.H. Degradation of epoxy polymers. 2. Mechanism of thermal degradation of bisphenol-A diglycidyl ether. Polym. Degrad. Stab. 1985, 13, 11–20. [Google Scholar] [CrossRef]
- Hutchinson, J.M.; Montserrat, S.; Román, F.; Cortés, P.; Campos, L. Intercalation of epoxy resin in organically modified montmorillonite. J. Appl. Polym. Sci. 2006, 102, 3751–3763. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the 2nd-order transitions of synthetic rubbers. 1. Non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Fox, T.G. Influence of diluent and of copolymer composition on the glass temperature of a polymer system. Bull. Am. Phys. Soc. 1956, 1, 123. [Google Scholar]
- Mandelkern, L.; Martin, G.M.; Quinn, F.A. Glassy state transitions of poly-(chlorotrifluoroethylene), poly-(vinylidenefluoride), and their copolymers. J. Res. Nat. Bur. Stand. 1957, 58, 137–143. [Google Scholar] [CrossRef]
- Pochan, J.M.; Beatty, C.L.; Pochan, D.F. Different approach for the correlation of the Tg of mixed amorphous systems. Polymer 1979, 20, 879–886. [Google Scholar] [CrossRef]
- Montserrat, S.; Román, F.; Hutchinson, J.M.; Campos, L. Analysis of the cure of epoxy based layered silicate nanocomposites: Reaction kinetics and nanostructure development. J. Appl. Polym. Sci. 2008, 108, 923–938. [Google Scholar] [CrossRef]
- Klute, C.H.; Viehmann, W. Heat of polymerization of phenyl glycidyl ether and of an epoxy resin. J. Appl. Polym. Sci. 1961, 5, 86–95. [Google Scholar] [CrossRef]
- Dell’Erba, I.E.; Williams, R.J.J. Homopolymerization of epoxy monomers initiated by 4-(dimethylamino)pyridine. Polym. Eng. Sci. 2006, 46, 351–359. [Google Scholar] [CrossRef]
- Kissinger, H.E. Variation of peak temperature with heating rate in differential thermal analysis. J. Res. Nat. Bur. Stand. 1956, 57, 217–221. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Cortés, P.; Fraga, I.; Calventus, Y.; Hutchinson, J.M. A New Epoxy Based Layered Silicate Nanocomposite Using a Hyperbranched Polymer: Study of the Curing Reaction and Nanostructure Development; Nanocomposites MoDeSt Workshop: Warsaw, Poland, 2013; pp. 91–94. [Google Scholar]
- Hutchinson, J.M. Physical aging of polymers. Prog. Polym. Sci. 1995, 20, 703–760. [Google Scholar] [CrossRef]
- Ngai, K.L.; Paluch, M. Classification of secondary relaxation in glass-formers based on dynamic properties. J. Chem. Phys. 2004, 120, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Turky, G.; Sangoro, J.R.; Abdel Rehim, M.; Kremer, F. Secondary relaxations and electrical conductivity in hyperbranched polyester amides. J. Polym. Sci. B Polym. Phys. 2010, 48, 1651–1657. [Google Scholar] [CrossRef]
- Prevosto, D.; Capaccioli, S.; Lucchesi, M.; Sharifi, S.; Kessairi, K.; Ngai, K.L. Relationship between structural and secondary relaxation in glass formers: Ratio between glass transition temperature and activation energy. Phil. Mag. 2008, 88, 4063–4069. [Google Scholar] [CrossRef]
- Serrano, E.; Kortaberria, G.; Arruti, P.; Tercjak, A.; Mondragon, I. Molecular dynamics of an epoxy resin modified with an epoxidized poly(styrene-butadiene) linear block copolymer during cure and microphase separation processes. Eur. Polym. J. 2009, 45, 1046–1057. [Google Scholar] [CrossRef]
- Mijovic, J.; Shen, M.; Sy, J.W. Dynamics and morphology in nanostructured thermoset network/block copolymer blends during network formation. Macromolecules 2000, 33, 5235–5244. [Google Scholar] [CrossRef]
- Hodge, I.M. Enthalpy relaxation and recovery in amorphous materials. J. Non-Cryst. Sol. 1994, 169, 211–266. [Google Scholar] [CrossRef]
- O’Reilly, J.M. Review of structure and mobility in amorphous polymers. CRC Crit. Rev. Solid State Mater. Sci. 1987, 13, 259–277. [Google Scholar] [CrossRef]
- Hodge, I.M. Effects of annealing and prior history on enthalpy relaxation in glassy polymers. 6. Adam-Gibbs formulation of non-linearity. Macromolecules 1987, 20, 2897–2908. [Google Scholar] [CrossRef]
- Hutchinson, J.M.; Ruddy, M. Thermal cycling of glasses. 2. Experimental evaluation of the structure (or nonlinearity) parameter x. J. Polym. Sci. B Polym. Phys. 1988, 26, 2341–2366. [Google Scholar] [CrossRef]
- Montserrat, S.; Cortés, P.; Pappin, A.J.; Quah, K.H.; Hutchinson, J.M. Structural relaxation in fully cured epoxy resins. J. Non-Cryst. Sol. 1994, 172–174, 1017–1022. [Google Scholar] [CrossRef]
- Hutchinson, J.M.; McCarthy, D.; Montserrat, S.; Cortés, P. Enthalpy relaxation in a partially cured epoxy resin. J. Polym. Sci. B Polym. Phys. 1996, 34, 229–239. [Google Scholar] [CrossRef]
- Montserrat, S.; Cortés, P.; Calventus, Y.; Hutchinson, J.M. Effect of crosslink length on the enthalpy relaxation of fully cured epoxy-diamine resins. J. Polym. Sci. B Polym. Phys. 2000, 38, 456–468. [Google Scholar] [CrossRef]
- Montserrat, S.; Román, F.; Colomer, P. Vitrification, devitrification, and dielectric relaxations during the non-isothermal curing of diepoxy-cycloaliphatic diamine. J. Appl. Polym. Sci. 2006, 102, 558–563. [Google Scholar] [CrossRef]
- Fraga, I.; Montserrat, S.; Hutchinson, J.M. TOPEM, a new temperature modulated DSC technique—Application to the glass transition of polymers. J. Therm. Anal. Calorim. 2007, 87, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Van Assche, G.; Van Hemelrijck, A.; Rahier, H.; Van Mele, B. Modulated differential scanning calorimetry: Non-isothermal cure, vitrification, and devitrification of thermosetting systems. Thermochim. Acta 1996, 286, 209–224. [Google Scholar] [CrossRef]
- Fraga, I.; Montserrat, S.; Hutchinson, J.M. Vitrification during the isothermal cure of thermosets. Part I. An investigation using TOPEM, a new temperature modulated technique. J. Therm. Anal. Calorim. 2008, 91, 687–695. [Google Scholar] [CrossRef]
- Fraga, I.; Montserrat, S.; Hutchinson, J.M. Vitrification and devitrification during the non-isothermal cure of a thermoset. Theoretical model and comparison with calorimetric experiments. Macromol. Chem. Phys. 2010, 211, 57–65. [Google Scholar] [CrossRef]
- Angell, C.A. Strong and fragile liquids. In Relaxations in Complex Systems; Ngai, K.L., Wright, G.B., Eds.; US Department of Commerce: Springfield, Virginia, 1984; pp. 3–11. [Google Scholar]
- Hutchinson, J.M. Interpretation of glass transition phenomena in the light of the strength-fragility concept. Polym. Int. 1998, 47, 56–64. [Google Scholar] [CrossRef]
- Fernandez-Francos, X.; Rybak, A.; Sekula, R.; Ramis, X.; Ferrando, F.; Okrasa, L.; Serra, A. Modification of epoxyanhydride thermosets with a hyperbranched poly(ester amide). II. Thermal, dynamic mechanical, and dielectric properties and thermal reworkability. J. Appl. Polym. Sci. 2013, 128, 4001–4013. [Google Scholar] [CrossRef]
- Tombari, E.; Salvetti, G.; Johari, G.P. The temperature and polymerization effects on the relaxation time and conductivity, and the evolution of the localized motions. J. Chem. Phys. 2000, 113, 6957–6965. [Google Scholar] [CrossRef]
- Román, F.; Hutchinson, J.M.; Colomer, P. Dielectric relaxation study of hyperbranched polymers of different molecular weight and their interaction with an epoxy resin. In Proceedings of the Central and Eastern European Committee for Thermal Analysis and Calorimetry (CEEC-TAC3), Ljubljana, Slovenia, 25–28 August 2015; p. 221.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Heating Rate (K/min) | T5% (°C) | Tonset (°C) | TDTGA (°C) |
|---|---|---|---|---|
| ELP | 2 | 303.1 | 316.8 | 342.3 |
| 10 | 305.1 | 346.9 | 373.6 | |
| Epoxy | 2 | 289.8 | 315.6 | 351.5 and 410.3 |
| 10 | 311.6 | 341.9 | 381.4 and 434.2 | |
| HBPEI | 2 | 272.0 | 351.7 | 369.4 |
| 10 | 301.2 | 379.1 | 398.8 |
| Sample | Heating Rate (K/min) | Tg0 (°C) | ΔH (J/g) | Tp (°C) | Tg∞a (°C) |
|---|---|---|---|---|---|
| HBPEI | 2 | −55.4 | |||
| 10 | −54.0 | ||||
| Epoxy | 2 | −17.2 | |||
| 10 | −16.0 | ||||
| ELP | 0.5 | −17.9 | 389 | 57.2 | 121.4 |
| 1 | −18.2 | 426 | 67.6 | 122.2 | |
| 2 | −18.0 | 409 | 76.3 | 124.8 | |
| 4 | −17.6 | 399 | 86.6 | 125.6 | |
| 10 | −15.2 | 317 | 98.1 | 126.2 | |
| ELP Cured plaque | 10 | 123.4 |
| Sample | Heating Rate (K/min) | DRS Relaxations | DMA Relaxations | |||
|---|---|---|---|---|---|---|
| γ | β | α | β | α | ||
| Ea (kJ/mol) | Ea (kJ/mol) | Eapp (kJ/mol) | Ea (kJ/mol) | Eapp (kJ/mol) | ||
| HBPEI | 0.5 | 42.0 | 86.4 | 78.1 | ||
| 2.0 | 41.0 | 98.9 | 63.3 | 99.0 | 209 | |
| Epoxy | 2.0 | 27.7 | 79.9 | 306 | 89.0 | 358 |
| ELP uncured | 0.5 | 36.0 | 88.0 | 302 | ||
| 1.0 | 36.3 | 92.4 | 297 | |||
| 2.0 | 27.3 | 74.3 | 263 | 79.0 | 344 | |
| 4.0 | 26.5 | 71.6 | 273 | |||
| Sample | Heating Rate (K/min) | DRS Relaxations | DMA Relaxations | ||
|---|---|---|---|---|---|
| β | α | β | α | ||
| Ea (kJ/mol) | Eapp (kJ/mol) | Ea(kJ/mol) | Eapp (kJ/mol) | ||
| cured ELP | 1 | 60.6 | 188 | ||
| 2 | 53.8 | 158 | 57.8 | 323 | |
| 5 | - | 172 | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Román, F.; Colomer, P.; Calventus, Y.; Hutchinson, J.M. Molecular Mobility in Hyperbranched Polymers and Their Interaction with an Epoxy Matrix. Materials 2016, 9, 192. https://doi.org/10.3390/ma9030192
Román F, Colomer P, Calventus Y, Hutchinson JM. Molecular Mobility in Hyperbranched Polymers and Their Interaction with an Epoxy Matrix. Materials. 2016; 9(3):192. https://doi.org/10.3390/ma9030192
Chicago/Turabian StyleRomán, Frida, Pere Colomer, Yolanda Calventus, and John M. Hutchinson. 2016. "Molecular Mobility in Hyperbranched Polymers and Their Interaction with an Epoxy Matrix" Materials 9, no. 3: 192. https://doi.org/10.3390/ma9030192