
Enhancement in Mechanical and Electrical Properties of Polypropylene Using Graphene Oxide Grafted with End-Functionalized Polypropylene

Abstract
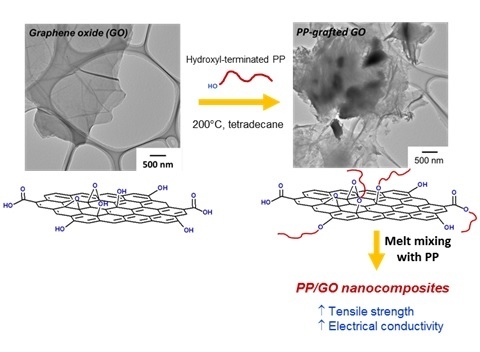
:
1. Introduction
2. Results and Discussion
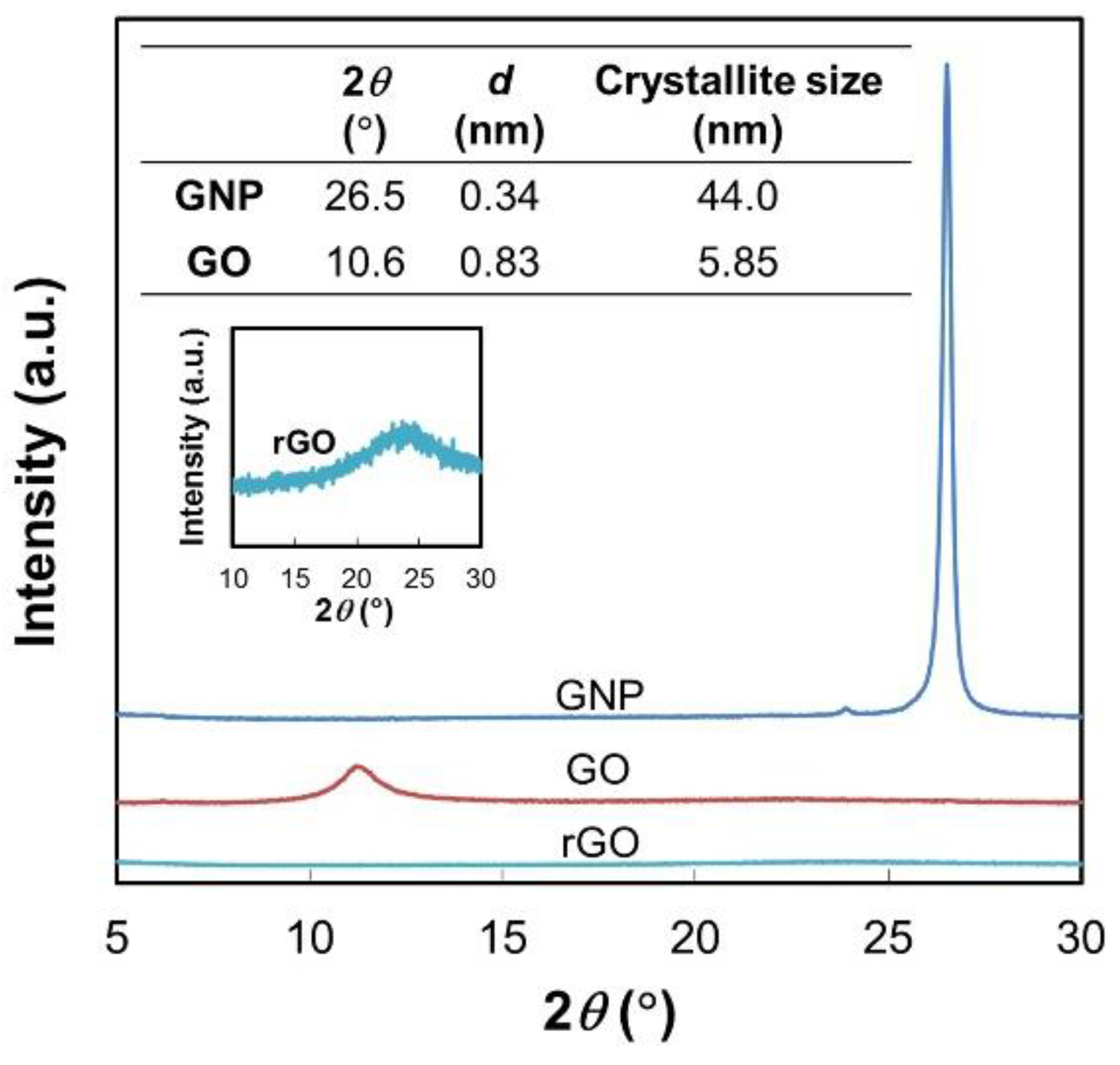
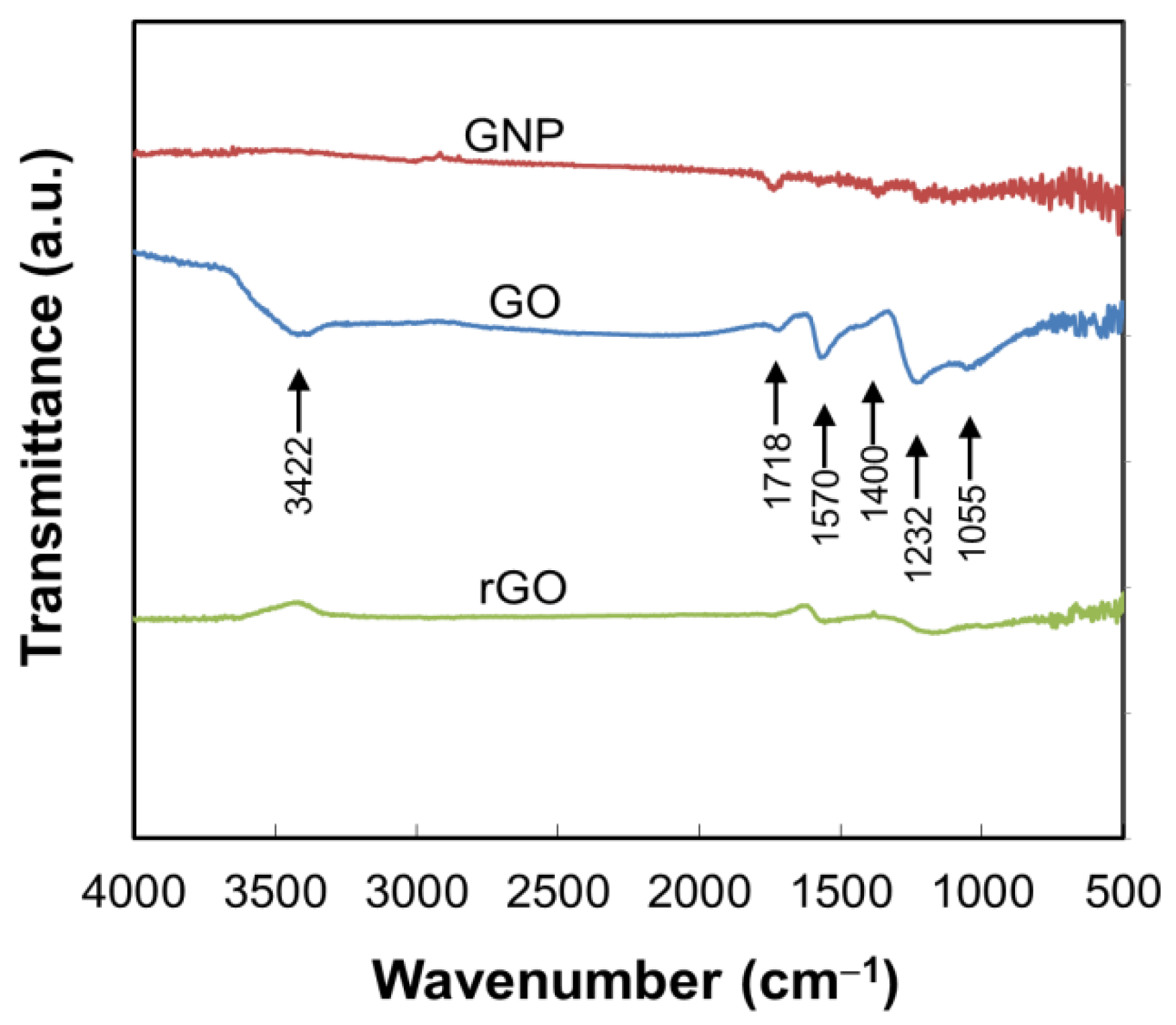
2.1. Preparation of GO and rGO
2.2. Synthesis of PP-OH
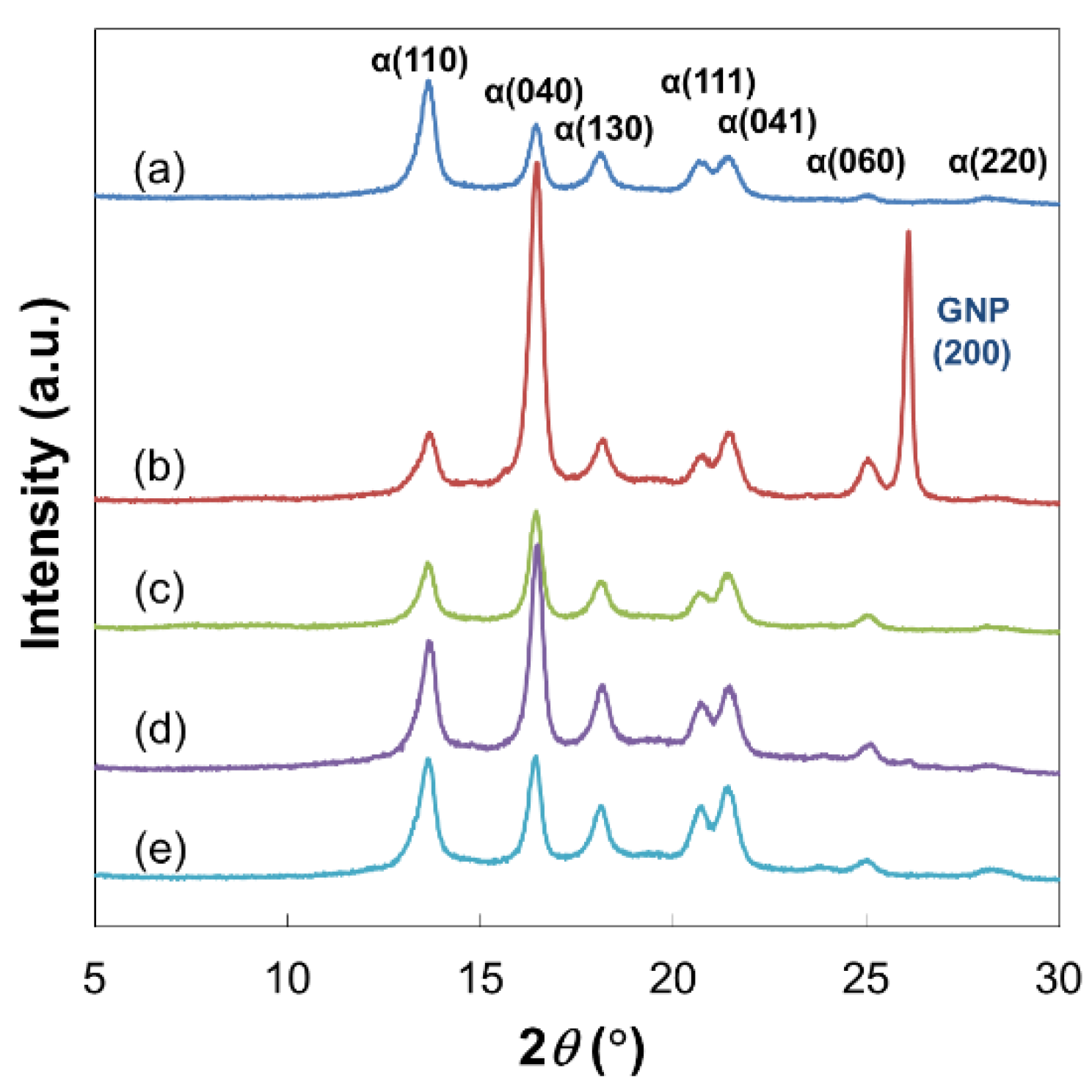
2.3. Preparation and Characterization of PP Nanocomposites
3. Experimental Section
3.1. Materials
3.2. Synthesis of PP-OH
3.3. Preparation of GO, rGO, and pGO
3.4. Preparation of PP-GO
3.5. Preparation of PP Nanocomposites
3.6. Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Takeuchi, K.; Terano, M.; Taniike, T. Sol-Gel Synthesis of Nano-Sized Silica in Confined Amorphous Space of Polypropylene: Impact of Nano-Level Structures of Silica on Physical Properties of Resultant Nanocomposites. Polymer 2014, 55, 1940–1947. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Stoller, M.D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R.S. Graphene-Based Ultracapacitors. Nano Lett. 2008, 8, 3498–3502. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Müller, M.B.; Gilje, S.; Kaner, R.B.; Wallace, G.G. Processable Aqueous Dispersions of Graphene Nanosheets. Nat. Nanotechnol. 2008, 3, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-Based Composite Materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wang, Y.; Huang, Y.; Ma, Y.; Liu, Z.; Cai, J.; Zhang, C.; Gao, H.; Chen, Y. Electromagnetic Interference Shielding of Graphene/Epoxy Composites. Carbon 2009, 47, 922–925. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Q.; Chen, D. Enhanced Mechanical Properties of Graphene-Based Poly(vinyl alcohol) Composites. Macromolecules 2010, 43, 2357–2363. [Google Scholar] [CrossRef]
- Kalaitzidou, K.; Fukushima, H.; Drzal, L.T. Mechanical Properties and Morphological Characterization of Exfoliated Graphite-Polypropylene Nanocomposites. Compos. Part A Appl. Sci. Manuf. 2007, 38, 1675–1682. [Google Scholar] [CrossRef]
- Achaby, M.E.; Arrakhiz, F.-E.; Vaudreuil, S.; Qaiss, A.K.; Bousmina, M.; Fassi-Fehri, O. Mechanical, Thermal, and Rheological Properties of Graphene-Based Polypropylene Nanocomposites Prepared by Melt Mixing. Polym. Compos. 2012, 33, 733–744. [Google Scholar] [CrossRef]
- Kalaitzidou, K.; Fukushima, H.; Drzal, L.T. A Route for Polymer Nanocomposites with Engineered Electrical Conductivity and Percolation Threshold. Materials 2010, 3, 1089–1103. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Brunner, P.J.; Masuda, J.; Hewlett, A.; Torkelson, J.M. Polypropylene-Graphite Nanocomposites Made by Solid-State Shear Pulverization: Effects of Significantly Exfoliated, Unmodified Graphite Content on Physical, Mechanical and Electrical Properties. Polymer 2010, 51, 5525–5531. [Google Scholar] [CrossRef]
- Song, P.; Cao, Z.; Cai, Y.; Zhao, L.; Fang, Z.; Fu, S. Fabrication of Exfoliated Graphene-Based Polypropylene Nanocomposites with Enhanced Mechanical and Thermal Properties. Polymer 2011, 52, 4001–4010. [Google Scholar] [CrossRef]
- Fu, S.; Li, N.; Wang, K.; Zhang, Q.; Fu, Q. Reduction of Graphene Oxide with the Presence of Polypropylene Micro-Latex for Facile Preparation of Polypropylene/Graphene Nanosheet Composites. Colloid Polym. Sci. 2015, 293, 1495–1503. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, X.; Zha, J.-W.; Zhao, J.; Dang, Z.-M.; Hu, G.-H. Dielectric Properties of Reduced Graphene Oxide/Polypropylene Composites with Ultralow Percolation Threshold. Polymer 2013, 54, 1916–1922. [Google Scholar] [CrossRef]
- Ryu, S.H.; Shanmugharaj, A.M. Influence of Long-Chain Alkylamine-Modified Graphene Oxide on the Crystallization, Mechanical and Electrical Properties of Isotactic Polypropylene Nanocomposites. Chem. Eng. J. 2014, 244, 552–560. [Google Scholar] [CrossRef]
- Ryu, S.H.; Shanmugharaj, A.M. Influence of Hexamethylene Diamine Functionalized Graphene Oxide on the Melt Crystallization and Properties of Polypropylene Nanocomposites. Mater. Chem. Phys. 2014, 146, 478–486. [Google Scholar] [CrossRef]
- Yuan, B.; Bao, C.; Song, L.; Hong, N.; Liew, K.M.; Hu, Y. Preparation of Functionalized Graphene Oxide/Polypropylene Nanocomposite with Significantly Improved Thermal Stability and Studies on the Crystallization Behavior and Mechanical Properties. Chem. Eng. J. 2014, 237, 411–420. [Google Scholar] [CrossRef]
- Song, N.; Yang, J.; Ding, P.; Tang, S.; Liu, Y.; Shi, L. Effect of Covalent-Functionalized Graphene Oxide with Polymer and Reactive Compatibilization on Thermal Properties of Maleic Anhydride Grafted Polypropylene. Ind. Eng. Chem. Res. 2014, 53, 19951–19960. [Google Scholar] [CrossRef]
- Taniike, T.; Toyonaga, M.; Terano, M. Polypropylene-Grafted Nanoparticles as a Promising Strategy for Boosting Physical Properties of Polypropylene-Based Nanocomposites. Polymer 2014, 55, 1012–1019. [Google Scholar] [CrossRef]
- Wang, Z.M.; Hong, H.; Chung, T.C. Synthesis of Maleic Anhydride Grafted Polypropylene with High Molecular Weight Using Borane/O2 Radical Initiator and Commercial PP Polymers. Macromolecules 2005, 38, 8966–8970. [Google Scholar] [CrossRef]
- Chung, T.C. Metallocene-Mediated Synthesis of Chain-End Functionalized Polypropylene and Application in PP/Clay Nanocomposites. J. Organomet. Chem. 2005, 690, 6292–6299. [Google Scholar] [CrossRef]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339–1339. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, J.; Wu, P. Polypropylene-Grafted Graphene Oxide Sheets as Multifunctional Compatibilizers for Polyolefin-Based Polymer Blends. J. Mater. Chem. 2012, 22, 14997–15005. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, J.; Wu, P. Preparation of Organically Dispersible Graphene Nanosheet Powders through a Lyophilization Method and their Poly(lactic acid) Composites. Carbon 2010, 48, 3834–3839. [Google Scholar] [CrossRef]
- Petit, C.; Bandosz, T.J. Graphite Oxide/Polyoxometalate Nanocomposites as Adsorbents of Ammonia. J. Phys. Chem. C 2009, 113, 3800–3809. [Google Scholar] [CrossRef]
- Aladekomo, J.B.; Bragg, R.H. Structural Transformations Induced in Graphite by Grinding: Analysis of 002 X-ray Diffraction Line Profiles. Carbon 1990, 2, 897–906. [Google Scholar] [CrossRef]
- Bragg, W.H.; Bragg, W.L. The Reflection of X-rays by Crystals. Proc. R. Soc. A 1913, 88, 428–438. [Google Scholar] [CrossRef]
- Dervishi, E.; Li, Z.; Watanabe, F.; Biswas, A.; Xu, Y.; Biris, A.R.; Saini, V.; Biris, A.S. Large-Scale Graphene Production by RF-cCVD Method. Chem. Commun. 2009, 27, 4061–4063. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Peng, T.; Sun, H.; Wang, J. The Effect of Thermal Exfoliation Temperature on the Structure and Supercapacitive Performance of Graphene Nanosheets. Nano-Micro Lett. 2015, 7, 17–26. [Google Scholar] [CrossRef]
- Huh, S.H. Thermal Reduction of Graphene Oxide. In Physics and Applications of Graphene–Experiments; Mikhailov, S., Ed.; InTech: Rijeka, Croatia, 2011; pp. 73–90. [Google Scholar]
- David, L.; Singh, G. Reduced Graphene Oxide Paper Electrode: Opposing Effect of Thermal Annealing on Li and Na Cyclability. J. Phys. Chem. C 2014, 118, 28401–28408. [Google Scholar] [CrossRef]
- Seredych, M.; Rossin, J.A.; Bandosz, T.J. Changes in Graphite Oxide Texture and Chemistry upon Oxidation and Reduction and their Effect on Adsorption of Ammonia. Carbon 2011, 49, 4392–4402. [Google Scholar] [CrossRef]
- Han, C.J.; Lee, M.S.; Byun, D.-J.; Kim, S.Y. Synthesis of Hydroxy-Terminated Polyethylene via Controlled Chain Transfer Reaction and Poly(ethylene-b-caprolactone) Block Copolymer. Macromolecules 2002, 35, 8923–8925. [Google Scholar] [CrossRef]
- Viana, M.M.; Lima, M.C.F.S.; Forsythe, J.C.; Gangoli, V.S.; Cho, M.; Cheng, Y.; Silva, G.G.; Wong, M.S.; Caliman, V. Facile Graphene Oxide Preparation by Microwave-Assisted Acid Method. J. Braz. Chem. Soc. 2015, 26. [Google Scholar] [CrossRef]
- Tu, N.D.K.; Choi, J.; Park, C.R.; Kim, H. Remarkable Conversion Between n- and p-Type Reduced Graphene Oxide on Varying the Thermal Annealing Temperature. Chem. Mater. 2015, 27, 7362–7369. [Google Scholar] [CrossRef]
- Acik, M.; Lee, G.; Mattevi, C.; Chhowalla, M.; Cho, K.; Chabal, Y.J. Unusual Infrared-Absorption Mechanism in Thermally Reduced Graphene Oxide. Nat. Mater. 2010, 9, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhai, W.; Tao, M.; Lu, D.; Zheng, W. Chemical Functionalization of Graphene Oxide Toward the Tailoring of the Interface in Polymer Composites. Compos. Sci. Technol. 2013, 77, 87–94. [Google Scholar] [CrossRef]
- Kalaitzidou, K.; Fukushima, H.; Askeland, P.; Drzal, L.T. The Nucleating Effect of Exfoliated Graphite Nanoplatelets and their Influence on the Crystal Structure and Electrical Conductivity of Polypropylene Nanocomposites. J. Mater. Sci. 2008, 43, 2895–2907. [Google Scholar] [CrossRef]
- Ferrage, E.; Martin, F.; Boudet, A.; Petit, S.; Fourty, G.; Jouffret, F.; Micoud, P.; de Parseval, P.; Salvi, S.; Bourgerette, C.; et al. Talc as Nucleating Agent of Polypropylene: Morphology Induced by Lamellar Particles Addition and Interface Mineral-Matrix Modelization. J. Mater. Sci. 2002, 37, 1561–1573. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | α(040)/α(110) |
|---|---|
| Pristine PP | 0.66 |
| PP/GNP (3.0 wt %) | 4.31 |
| PP/rGO (3.0 wt %) | 1.93 |
| PP/pGO (3.0 wt %) | 1.68 |
| PP/PP-GO (3.0 wt %) | 1.02 |
| Sample | t1/2−1 a (min−1) | Xc b (%) | Tm c (°C) |
|---|---|---|---|
| Pristine PP | 0.18 | 47.9 | 164 |
| PP/GNP (1.0 wt %) | 0.68 | 47.1 | 161 |
| PP/GNP (3.0 wt %) | 0.97 | 46.0 | 161 |
| PP/rGO (1.0 wt %) | 0.74 | 47.6 | 162 |
| PP/rGO (3.0 wt %) | 1.03 | 46.5 | 163 |
| PP/PP-GO (1.0 wt %) | 0.35 | 46.5 | 159 |
| PP/PP-GO (3.0 wt %) | 0.40 | 47.1 | 162 |
| Sample | Tensile Strength (MPa) | Young’s Modulus (MPa) | Elongation at Break (%) |
|---|---|---|---|
| Pristine PP | 30.2 ± 0.4 | 470 ± 15 | >300 |
| PP/GNP (1.0 wt % ) | 36.3 ± 1.1 | 550 ± 22 | >300 |
| PP/GNP (3.0 wt %) | 35.1 ± 0.7 | 567 ± 14 | 38 ± 8 |
| PP/rGO (1.0 wt %) | 36.4 ± 0.4 | 554 ± 24 | >300 |
| PP/rGO (3.0 wt %) | 36.2 ± 0.6 | 552 ± 40 | 62 ± 14 |
| PP/pGO (1.0 wt %) | 35.3 ± 0.6 | 516 ± 29 | >300 |
| PP/pGO (3.0 wt %) | 35.3 ± 0.5 | 549 ± 25 | 79 ± 12 |
| PP/PP-GO (1.0 wt %) | 38.7 ± 1.0 | 562 ± 34 | >300 |
| PP/PP-GO (3.0 wt % ) | 37.1 ± 0.5 | 564 ± 47 | >300 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chammingkwan, P.; Matsushita, K.; Taniike, T.; Terano, M. Enhancement in Mechanical and Electrical Properties of Polypropylene Using Graphene Oxide Grafted with End-Functionalized Polypropylene. Materials 2016, 9, 240. https://doi.org/10.3390/ma9040240
Chammingkwan P, Matsushita K, Taniike T, Terano M. Enhancement in Mechanical and Electrical Properties of Polypropylene Using Graphene Oxide Grafted with End-Functionalized Polypropylene. Materials. 2016; 9(4):240. https://doi.org/10.3390/ma9040240
Chicago/Turabian StyleChammingkwan, Patchanee, Katsuhiko Matsushita, Toshiaki Taniike, and Minoru Terano. 2016. "Enhancement in Mechanical and Electrical Properties of Polypropylene Using Graphene Oxide Grafted with End-Functionalized Polypropylene" Materials 9, no. 4: 240. https://doi.org/10.3390/ma9040240