
Si96: A New Silicon Allotrope with Interesting Physical Properties


Abstract
:1. Introduction
2. Materials and Methods
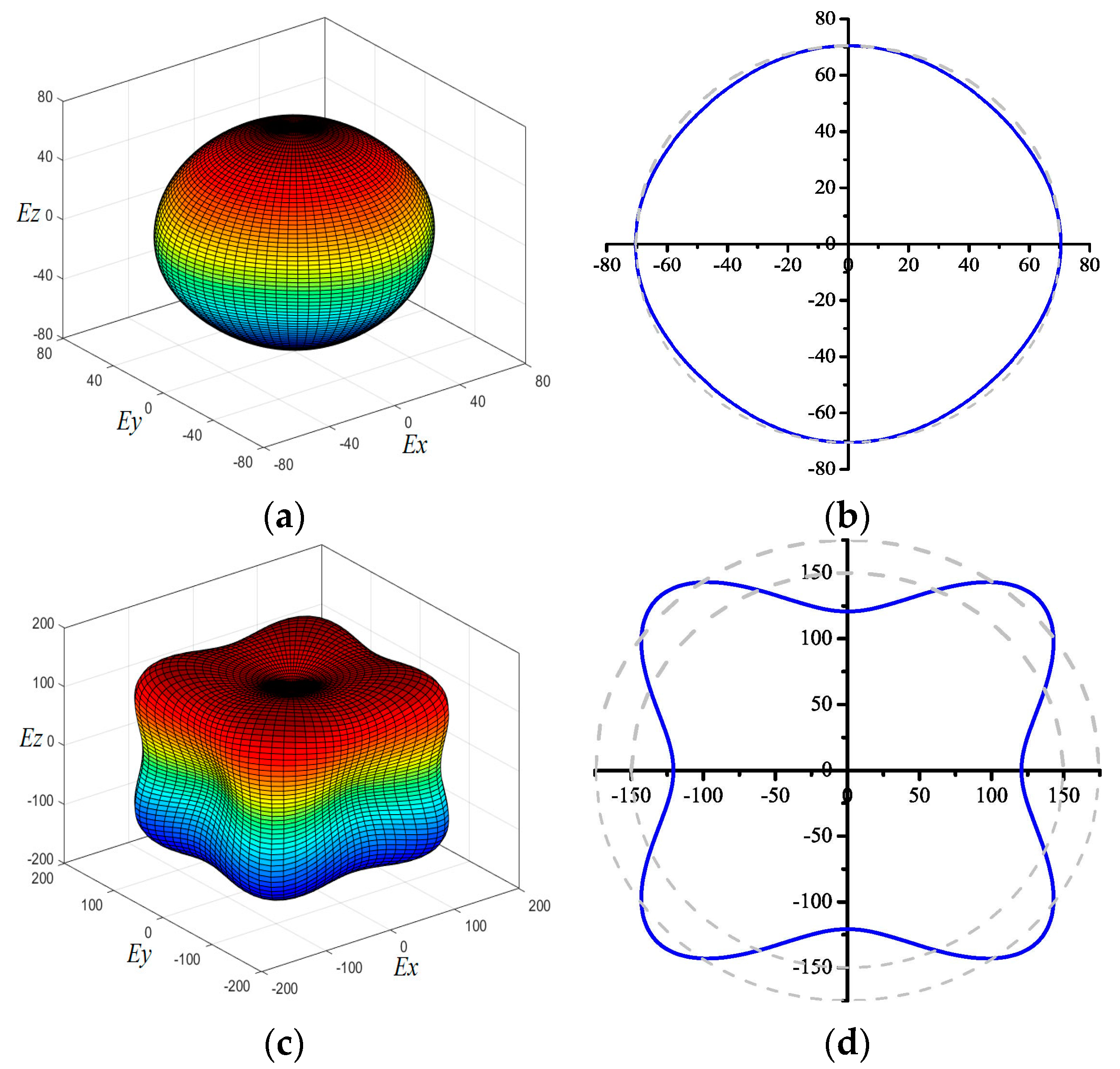
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mujica, A.; Rubio, A.; Munoz, A.; Needs, R.J. High-pressure phases of group-IV, III-V, and II-VI compounds. Rev. Mod. Phys. 2003, 75, 863–912. [Google Scholar] [CrossRef]
- Bautista-Hernandez, A.; Rangel, T.; Romero, A.H.; Rignanese, G.M.; Salazar-Villanueva, M.; Chigo-Anota, E. Structural and vibrational stability of M and Z phases of silicon and germanium from first principles. J. Appl. Phys. 2013, 113, 193504. [Google Scholar] [CrossRef]
- Amsler, M.; Flores-Livas, J.A.; Lehtovaara, L.; Balima, F.; Ghasemi, S.A.; Machon, D.; Pailhes, S.; Willand, A.; Caliste, D.; Botti, S.; et al. Crystal Structure of Cold Compressed Graphite. Phys. Rev. Lett. 2012, 108, 065501. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Koretsune, T.; Saito, S.; Miyake, T.; Oshiyama, A. A new crystalline phase of four-fold coordinated silicon and germanium. New J. Phys. 2008, 10, 083001. [Google Scholar] [CrossRef]
- Wu, F.; Jun, D.; Kan, E.J.; Li, Z.Y. Density functional predictions of new silicon allotropes: Electronic properties and potential applications to Li-battery anode materials. Solid State Commun. 2011, 151, 1228–1230. [Google Scholar] [CrossRef]
- De Amrit Pryor, C.E. Electronic structure and optical properties of Si, Ge and diamond in the lonsdaleite phase. J. Phys. Condens. Matter 2014, 26, 045801. [Google Scholar] [CrossRef]
- Zhao, Z.S.; Tian, F.; Dong, X.; Li, Q.; Wang, Q.Q.; Wang, H.; Zhong, X.; Xu, B.; Yu, D.; He, J.L.; et al. Tetragonal allotrope of group 14 Elements. J. Am. Chem. Soc. 2012, 134, 12362–12365. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.D.; Sau, J.D.; Cohen, M.L. Ab initio survey of the electronic structure of tetrahedrally bonded phases of silicon. Phys. Rev. B 2008, 78, 035210. [Google Scholar] [CrossRef]
- Zhao, Z.S.; Xu, B.; Zhou, X.F.; Wang, L.M.; Wen, B.; He, J.L.; Liu, Z.Y.; Wang, H.T.; Tian, Y.J. Novel Superhard Carbon: C-Centered Orthorhombic C8. Phys. Rev. Lett. 2011, 107, 215502. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yan, H.Y.; Zhao, Y.B.; Yang, Y.T.; Yu, X.H.; Liu, Y.; Xing, M.J.; Zhang, J.Q.; et al. Novel silicon allotropes: Stability, mechanical, and electronic properties. J. Appl. Phys. 2015, 118, 185704. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T.; Yang, Q.; Chen, P.Y.; Xing, M.J.; Zhang, J.Q.; Yao, R.H. Prediction of novel phase of silicon and Si–Ge alloys. J. Solid State Chem. 2016, 233, 471–483. [Google Scholar] [CrossRef]
- Xiang, H.J.; Huang, B.; Kan, E.J.; Wei, S.H.; Gong, X.G. Towards direct-gap silicon phases by the inverse band structure design approach. Phys. Rev. Lett. 2013, 110, 118702. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Q.; Xu, B.; Sun, J.; Liu, H.Y.; Zhao, Z.S.; Yu, D.L.; Fan, C.Z.; He, J.L. Direct band gap silicon allotropes. J. Am. Chem. Soc. 2014, 136, 9826–9829. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Cote, M.; Louie, S.G.; Cohen, M.L. Ab initio study of silicon in the R8 phase. Phys. Rev. B 1997, 56, 6662. [Google Scholar] [CrossRef]
- Malone, B.D.; Cohen, M.L. Prediction of a metastable phase of silicon in the Ibam structure. Phys. Rev. B 2012, 85, 024116. [Google Scholar] [CrossRef]
- Karttunen, A.J.; Fassler, T.F.; Linnolahti, M.; Pakkanen, T.A. Structural principles of semiconducting group 14 clathrate frameworks. Inorg. Chem. 2011, 50, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Zwijnenburg, M.A.; Jelfsab, K.E.; Bromley, S.T. An extensive theoretical survey of low-density allotropy in silicon. Phys. Chem. Chem. Phys. 2010, 12, 8505–8512. [Google Scholar] [CrossRef] [PubMed]
- Amsler, M.; Botti, S.; Marques, M.A.L.; Lenosky, T.J.; Goedecker, S. Low-density silicon allotropes for photovoltaic applications. Phys. Rev. B 2015, 92, 014101. [Google Scholar] [CrossRef]
- Li, D.; Tian, F.B.; Chu, B.H.; Duan, D.F.; Wei, S.L.; Lv, Y.Z.; Zhang, H.D.; Wang, L.; Lu, N.; Liu, B.B.; et al. Cubic C96: A novel carbon allotrope with a porous nanocube network. J. Mater. Chem. A 2015, 3, 10448–10452. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-Newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; Gironcoli, S.; de Corso, A.; dal Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–564. [Google Scholar] [CrossRef]
- Lyakhov, A.O.; Oganov, A.R. Evolutionary search for superhard materials: Methodology and applications to forms of carbon and TiO2. Phys. Rev. B 2011, 84, 092103. [Google Scholar] [CrossRef]
- Gilman, J.J. Flow of covalent solids at low temperatures. J. Appl. Phys. 1975, 46, 5110–5113. [Google Scholar] [CrossRef]
- Lawn, B.R.; Evans, A.G.; Marshall, D.B. Elastic/Plastic Indentation Damage in Ceramics: The Median/Radial Crack System. J. Am. Ceram. Soc. 1980, 63, 574–581. [Google Scholar] [CrossRef]
- Danyluk, S.; Lim, D.S.; Kalejs, J. Microhardness of carbon-doped (111) p-type Czochralski silicon. J. Mater. Sci. Lett. 1985, 4, 1135–1137. [Google Scholar] [CrossRef]
- Feltham, P.; Banerjee, R. Theory and application of microindentation in studies of glide and cracking in single crystals of elemental and compound semiconductors. J. Mater. Sci. 1992, 27, 1626–1632. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Wei, Q.; Yan, H.Y.; Zhang, M.G.; Zhang, D.Y.; Zhang, J.Q. A New Potential Superhard Phase of OsN2. Acta Phys. Pol. A 2014, 126, 740–746. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philosop. Mag. J. Sci. Ser. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Basile, G.; Bergamin, A.; Cavagenro, G.; Mana, G.; Vittone, E.; Zosi, G. Measurement of the silicon (220) lattice spacing. Phys. Rev. Lett. 1994, 72, 3133–3136. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S. Group-IV semiconductors. In Handbook on Physical Properties of Semiconductors; Kluwer Academic Publishers: Boston, MA, USA, 2004; Volume 2, p. 46. [Google Scholar]
- Gomez-Abal, R.; Li, X.; Scheffler, M.; Ambrosch-Draxl, C. Influence of the core-valence interaction and of the pseudopotential approximation on the electron self-energy in semiconductors. Phys. Rev. Lett. 2008, 101, 106404. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Erratum: “Hybrid functionals based on a screened Coulomb potential” [J. Chem. Phys. 118, 8207 (2003)]. J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Duan, Y.H.; Sun, Y.; Peng, M.J.; Zhou, S.G. Anisotropic elastic properties of the Ca–Pb compounds. J. Alloys Compd. 2014, 595, 14–21. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comput. Mater. Sci. 2014, 83, 27–34. [Google Scholar] [CrossRef]
- Jung, S.C.; Han, Y.K. Ab initio molecular dynamics simulation of lithiation-induced phase-transition of crystalline silicon. Electrochim. Acta 2012, 62, 73–76. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Work | a | ρ | C11 | C12 | C44 | B | G | B/G | E | v |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Si96 | This work | 13.710 | 1.737 | 89 | 33 | 26 | 52 | 27 | 1.93 | 69 | 0.28 |
| Diamond Si | This work | 5.436 | 2.322 | 165 | 65 | 87 | 98 | 70 | 1.40 | 170 | 0.21 |
| Diamond Si | Experimental | 5.431 1 | 2.329 2 | 166 3 | 64 | 80 | 102 | - | - | - | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Chai, C.; Wei, Q.; Zhou, P.; Zhang, J.; Yang, Y. Si96: A New Silicon Allotrope with Interesting Physical Properties. Materials 2016, 9, 284. https://doi.org/10.3390/ma9040284
Fan Q, Chai C, Wei Q, Zhou P, Zhang J, Yang Y. Si96: A New Silicon Allotrope with Interesting Physical Properties. Materials. 2016; 9(4):284. https://doi.org/10.3390/ma9040284
Chicago/Turabian StyleFan, Qingyang, Changchun Chai, Qun Wei, Peikun Zhou, Junqin Zhang, and Yintang Yang. 2016. "Si96: A New Silicon Allotrope with Interesting Physical Properties" Materials 9, no. 4: 284. https://doi.org/10.3390/ma9040284