
Poly(lactide)-g-poly(butylene succinate-co-adipate) with High Crystallization Capacity and Migration Resistance



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Crotonic Acid Functionalized PLA
2.3. PBSA Plasticizer Coupled PLA
2.4. Film Formation
2.5. Hydrolytic Degradation
2.6. Characterization of PLA, PLA/PBSA and PLA-CA-PBSA
3. Results and Discussion
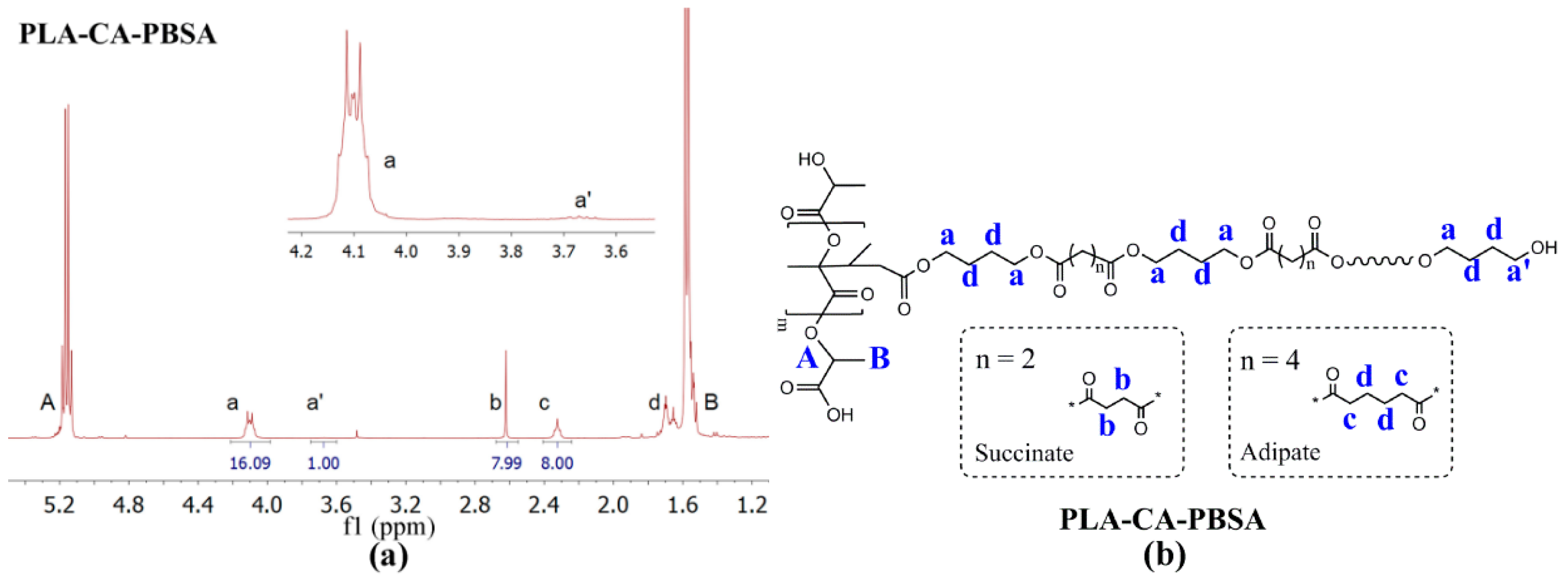
3.1. Synthesis of PLA-CA-PBSA
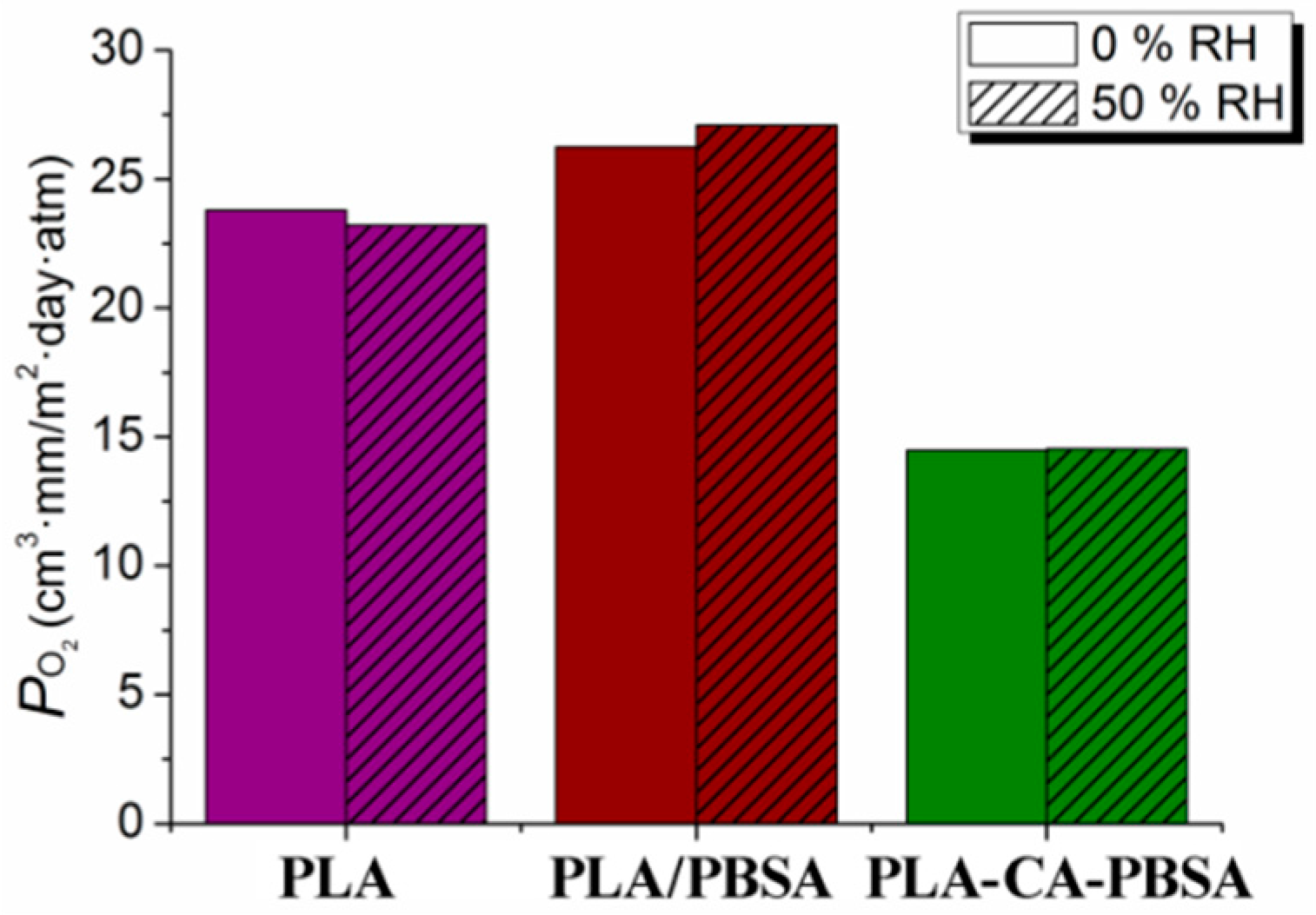
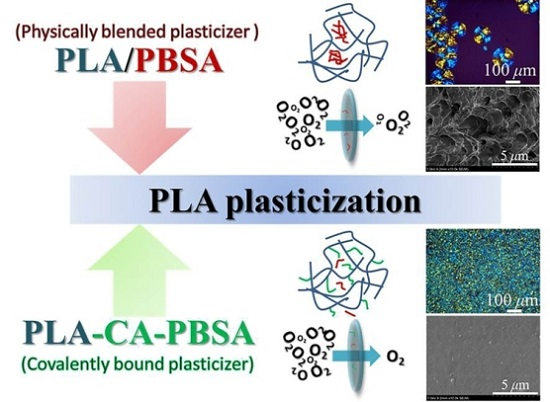
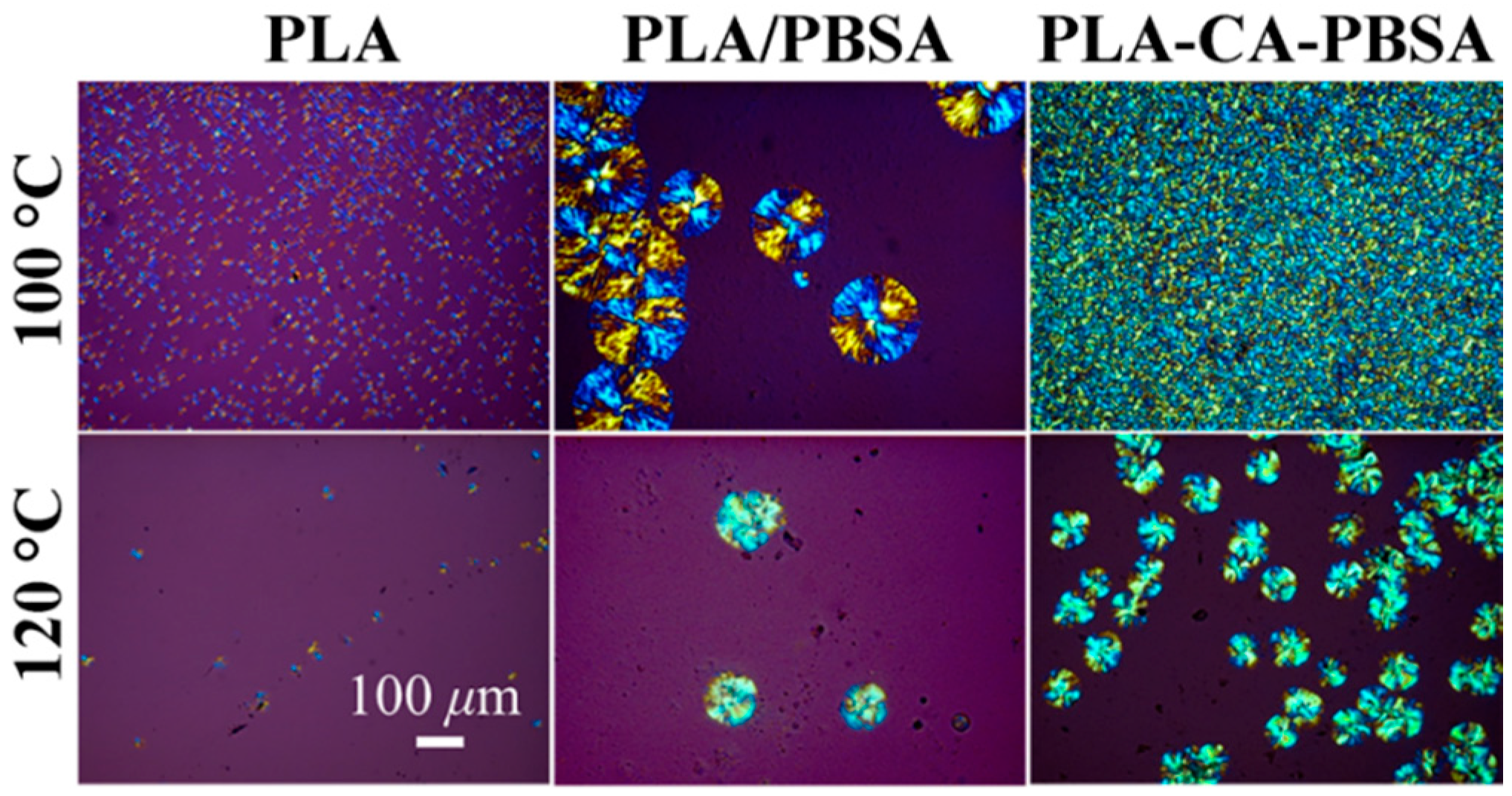
3.2. Characterization of Compression Molded Film PLA, PLA/PBSA and PLA-CA-PBSA
3.3. Hydrolytic Degradation and Migration Test of Compression Molded Film PLA, PLA/PBSA and PLA-CA-PBSA
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PLA | polylactide |
| PBSA | poly(butylene succinate-co-adipate) |
| CA | crotonic acid |
| PEG | poly(ethylene glycol) |
| Acrylated PEG | acrylated poly(ethylene glycol) |
| PHB | poly(3-hydroxybutyrate) |
| PBA | poly(butylene adipate) |
| PBS | poly(butylene succinate) |
| TPP | triphenyl phosphite |
| TIP | titanium isopropoxide |
| PLA-CA | crotonic acid grafted PLA |
| PLA/PBSA | PLA/PBSA blends |
| PLA-CA-PBSA | PBSA coupled PLA-CA |
References
- Martin, O.; Avérous, L. Poly(lactic acid): Plasticization and properties of biodegradable multiphase systems. Polymer 2001, 42, 6209–6219. [Google Scholar] [CrossRef]
- Xiao, H.; Lu, W.; Yeh, J.-T. Effect of plasticizer on the crystallization behavior of poly(lactic acid). J. Appl. Polym. Sci. 2009, 113, 112–121. [Google Scholar] [CrossRef]
- Chieng, B.W.; Ibrahim, N.A.; Yunus, W.M.Z.W.; Hussein, M.Z. Plasticized poly(lactic acid) with low molecular weight poly(ethylene glycol): Mechanical, thermal, and morphology properties. J. Appl. Polym. Sci. 2013, 130, 4576–4580. [Google Scholar] [CrossRef]
- Liao, R.; Yang, B.; Yu, W.; Zhou, C. Isothermal cold crystallization kinetics of polylactide/nucleating agents. J. Appl. Polym. Sci. 2007, 104, 310–317. [Google Scholar] [CrossRef]
- Xu, Z.; Niu, Y.; Wang, Z.; Li, H.; Yang, L.; Qiu, J.; Wang, H. Enhanced nucleation rate of polylactide in composites assisted by surface acid oxidized carbon nanotubes of different aspect ratios. ACS Appl. Mater. Interfaces 2011, 3, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Feng, Z.-X.; Xie, L.; Hakkarainen, M. Graphene oxide-driven design of strong and flexible biopolymer barrier films: From smart crystallization control to affordable engineering. ACS Sustain. Chem. Eng. 2016, 4, 334–349. [Google Scholar] [CrossRef]
- Xu, H.; Wu, D.; Yang, X.; Xie, L.; Hakkarainen, M. Thermostable and impermeable “nano-barrier walls” constructed by poly(lactic acid) stereocomplex crystal decorated graphene oxide nanosheets. Macromolecules 2015, 48, 2127–2137. [Google Scholar] [CrossRef]
- Schmidt, S.C.; Hillmyer, M.A. Polylactide stereocomplex crystallites as nucleating agents for isotactic polylactide. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 300–313. [Google Scholar] [CrossRef]
- Tachibana, Y.; Maeda, T.; Ito, O.; Maeda, Y.; Kunioka, M. Biobased myo-inositol as nucleator and stabilizer for poly(lactic acid). Polym. Degrad. Stab. 2010, 95, 1321–1329. [Google Scholar] [CrossRef]
- Wang, L.; Jing, X.; Cheng, H.; Hu, X.; Yang, L.; Huang, Y. Rheology and crystallization of long-chain branched poly(l-lactide)s with controlled branch length. Ind. Eng. Chem. Res. 2012, 51, 10731–10741. [Google Scholar] [CrossRef]
- Kfoury, G.; Raquez, J.-M.; Hassouna, F.; Leclère, P.; Toniazzo, V.; Ruch, D.; Dubois, P. Toughening of poly(lactide) using polyethylene glycol methyl ether acrylate: Reactive versus physical blending. Polym. Eng. Sci. 2015, 55, 1408–1419. [Google Scholar] [CrossRef]
- Choi, K.; Choi, M.-C.; Han, D.-H.; Park, T.-S.; Ha, C.-S. Plasticization of poly(lactic acid) (PLA) through chemical grafting of poly(ethylene glycol) (PEG) via in situ reactive blending. Eur. Polym. J. 2013, 49, 2356–2364. [Google Scholar] [CrossRef]
- Cicero, J.A.; Dorgan, J.R.; Garrett, J.; Runt, J.; Lin, J.S. Effects of molecular architecture on two-step, melt-spun poly(lactic acid) fibers. J. Appl. Polym. Sci. 2002, 86, 2839–2846. [Google Scholar] [CrossRef]
- Carlson, D.; Nie, L.; Narayan, R.; Dubois, P. Maleation of polylactide (PLA) by reactive extrusion. J. Appl. Polym. Sci. 1999, 72, 477–485. [Google Scholar] [CrossRef]
- Höglund, A.; Hakkarainen, M.; Edlund, U.; Albertsson, A.-C. Surface modification changes the degradation process and degradation product pattern of polylactide. Langmuir 2009, 26, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.W.; Li, P.; Ren, X.; Jiang, T.; Yeh, J.-T. Isothermal crystallization kinetics and crystal structure of poly(lactic acid): Effect of triphenyl phosphate and talc. J. Appl. Polym. Sci. 2010, 118, 3558–3569. [Google Scholar] [CrossRef]
- Li, H.; Huneault, M.A. Effect of nucleation and plasticization on the crystallization of poly(lactic acid). Polymer 2007, 48, 6855–6866. [Google Scholar] [CrossRef]
- Yang, X.; Clénet, J.; Xu, H.; Odelius, K.; Hakkarainen, M. Two step extrusion process: From thermal recycling of PHB to plasticized PLA by reactive extrusion grafting of PHB degradation products onto PLA chains. Macromolecules 2015, 48, 2509–2518. [Google Scholar] [CrossRef]
- Jompang, L.; Thumsorn, S.; On, J.W.; Surin, P.; Apawet, C.; Chaichalermwong, T.; Kaabbuathong, N.; O-Charoen, N.; Srisawat, N. Poly(lactic acid) and poly(butylene succinate) blend fibers prepared by melt spinning technique. Energy Procedia 2013, 34, 493–499. [Google Scholar] [CrossRef]
- Ojijo, V.; Sinha Ray, S.; Sadiku, R. Role of specific interfacial area in controlling properties of immiscible blends of biodegradable polylactide and poly[(butylene succinate)-co-adipate]. ACS Appl. Mater. Interfaces 2012, 4, 6690–6701. [Google Scholar] [CrossRef] [PubMed]
- Pivsa-Art, S.; Thumsorn, S.; Pavasupree, S.; O-Charoen, N.; Pivsa-Art, W.; Yamane, H.; Ohara, H. Effect of additive on crystallization and mechanical properties of polymer blends of poly(lactic acid) and poly[(butylene succinate)-co-adipate]. Energy Procedia 2013, 34, 563–571. [Google Scholar] [CrossRef]
- Yokohara, T.; Yamaguchi, M. Structure and properties for biomass-based polyester blends of PLA and PBS. Eur. Polym. J. 2008, 44, 677–685. [Google Scholar] [CrossRef]
- Pivsa-Art, W.; Pavasupree, S.; O-Charoen, N.; Insuan, U.; Jailak, P.; Pivsa-Art, S. Preparation of polymer blends between poly (l-lactic acid), poly (butylene succinate-co-adipate) and poly (butylene adipate-co-terephthalate) for blow film industrial application. Energy Procedia 2011, 9, 581–588. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.W. Characterization and processing of biodegradable polymer blends of poly (lactic acid) with poly (butylene succinate adipate). Korea Aust. Rheol. J. 2005, 17, 71–77. [Google Scholar]
- Fabbri, M.; Soccio, M.; Costa, M.; Lotti, N.; Gazzano, M.; Siracusa, V.; Gamberini, R.; Rimini, B.; Munari, A.; García-Fernández, L.; et al. New fully bio-based PLLA triblock copoly(ester urethane)s as potential candidates for soft tissue engineering. Polym. Degrad. Stab. 2016. [Google Scholar] [CrossRef]
- Ojijo, V.; Cele, H.; Sinha Ray, S. Morphology and properties of polymer composites based on biodegradable polylactide/poly[(butylene succinate)-co-adipate] blend and nanoclay. Macromol. Mater. Eng. 2011, 296, 865–877. [Google Scholar] [CrossRef]
- Ojijo, V.; Sinha Ray, S.; Sadiku, R. Toughening of biodegradable polylactide/poly(butylene succinate-co-adipate) blends via in situ reactive compatibilization. ACS Appl. Mater. Interfaces 2013, 5, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hakkarainen, M. Migration resistant glucose esters as bioplasticizers for polylactide. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Lindström, A.; Hakkarainen, M. Designed chain architecture for enhanced migration resistance and property preservation in poly(vinyl chloride)/polyester blends. Biomacromolecules 2007, 8, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Lindström, A.; Hakkarainen, M. Environmentally friendly plasticizers for poly(vinyl chloride)—Improved mechanical properties and compatibility by using branched poly(butylene adipate) as a polymeric plasticizer. J. Appl. Polym. Sci. 2006, 100, 2180–2188. [Google Scholar] [CrossRef]
- Siracusa, V.; Lotti, N.; Munari, A.; Dalla Rosa, M. Poly(butylene succinate) and poly(butylene succinate-co-adipate) for food packaging applications: Gas barrier properties after stressed treatments. Polym. Degrad. Stab. 2015, 119, 35–45. [Google Scholar] [CrossRef]
- Yang, X.; Odelius, K.; Hakkarainen, M. Microwave-assisted reaction in green solvents recycles PHB to functional chemicals. ACS Sustain. Chem. Eng. 2014, 2, 2198–2203. [Google Scholar] [CrossRef]
- Park, J.W.; Im, S.S. Phase behavior and morphology in blends of poly(l-lactic acid) and poly(butylene succinate). J. Appl. Polym. Sci. 2002, 86, 647–655. [Google Scholar] [CrossRef]
- Shibata, M.; Inoue, Y.; Miyoshi, M. Mechanical properties, morphology, and crystallization behavior of blends of poly(l-lactide) with poly(butylene succinate-co-l-lactate) and poly(butylene succinate). Polymer 2006, 47, 3557–3564. [Google Scholar] [CrossRef]
- Odelius, K.; Ohlson, M.; Höglund, A.; Albertsson, A.-C. Polyesters with small structural variations improve the mechanical properties of polylactide. J. Appl. Polym. Sci. 2013, 127, 27–33. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, S.; Zhang, L.; Bai, Y. Crystallization behavior of long-chain branching polylactide. Ind. Eng. Chem. Res. 2012, 51, 13670–13679. [Google Scholar] [CrossRef]
- Nouri, S.; Dubois, C.; Lafleur, P.G. Homocrystal and stereocomplex formation behavior of polylactides with different branched structures. Polymer 2015, 67, 227–239. [Google Scholar] [CrossRef]
- Xu, H.; Xie, L.; Jiang, X.; Hakkarainen, M.; Chen, J.-B.; Zhong, G.-J.; Li, Z.-M. Structural basis for unique hierarchical cylindrites induced by ultrahigh shear gradient in single natural fiber reinforced poly(lactic acid) green composites. Biomacromolecules 2014, 15, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Yan, C.; Lu, J.; Yang, W. Miscible crystalline/crystalline polymer blends of poly(vinylidene fluoride) and poly(butylene succinate-co-butylene adipate): Spherulitic morphologies and crystallization kinetics. Macromolecules 2007, 40, 5047–5053. [Google Scholar] [CrossRef]
- Zhang, J.; Tashiro, K.; Tsuji, H.; Domb, A.J. Disorder-to-order phase transition and multiple melting behavior of poly(l-lactide) investigated by simultaneous measurements of WAXD and DSC. Macromolecules 2008, 41, 1352–1357. [Google Scholar] [CrossRef]
- Guinault, A.; Sollogoub, C.; Ducruet, V.; Domenek, S. Impact of crystallinity of poly(lactide) on helium and oxygen barrier properties. Eur. Polym. J. 2012, 48, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Cocca, M.; Lorenzo, M.L.D.; Malinconico, M.; Frezza, V. Influence of crystal polymorphism on mechanical and barrier properties of poly(l-lactic acid). Eur. Polym. J. 2011, 47, 1073–1080. [Google Scholar] [CrossRef]
- Delpouve, N.; Stoclet, G.; Saiter, A.; Dargent, E.; Marais, S. Water barrier properties in biaxially drawn poly(lactic acid) films. J. Phys. Chem. B 2012, 116, 4615–4625. [Google Scholar] [CrossRef] [PubMed]
- Drieskens, M.; Peeters, R.; Mullens, J.; Franco, D.; Lemstra, P.J.; Hristova-Bogaerds, D.G. Structure versus properties relationship of poly(lactic acid). I. Effect of crystallinity on barrier properties. J. Polym. Sci. Part B Polym. Phys. 2009, 47, 2247–2258. [Google Scholar] [CrossRef]
- Bai, H.; Huang, C.; Xiu, H.; Zhang, Q.; Deng, H.; Wang, K.; Chen, F.; Fu, Q. Significantly improving oxygen barrier properties of polylactide via constructing parallel-aligned shish-kebab-like crystals with well-interlocked boundaries. Biomacromolecules 2014, 15, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.; Wyser, Y. Recent innovations in barrier technologies for plastic packaging—A review. Packag. Technol. Sci. 2003, 16, 149–158. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn (g/mol) | Mw (g/mol) | Mz (g/mol) | Ð |
|---|---|---|---|---|
| PLA | 160,000 | 279,000 | 411,000 | 1.7 |
| PLA-CA | 150,000 | 257,000 | 379,000 | 1.7 |
| PLA/PBSA | 138,000/1900 | 254,000/6700 | 388,000/13,300 | 1.8/3.4 |
| PLA-CA-PBSA | 181,000/11,900 | 279,000/17,200 | 387,000/23,200 | 1.5/1.4 |
| Sample | Tcc * (°C) | Tm * (°C) | Χc * (%) | Tmc ** (°C) | |
| PLA | - | 142.9 ± 0.2 | 2.1 ± 0.2 | - | |
| PLA/PBSA | 79.3 ± 0.8 | 133.0 ± 1.2 | 145.2 ± 0.2 | 8.4 ± 0.3 | 83.1 ± 1.2 |
| PLA-CA-PBSA | 76.3 ± 0.2 | 131.1 ± 0.2 | 145.1 ± 0.0 | 10.6 ± 1.0 | 80.4 ± 0.3 |
| Sample | Tg *** (°C) | Tcc *** (°C) | Tm *** (°C) | ||
| PLA | 44.3 ± 0.7 | - | 142.2 ± 0.1 | ||
| PLA/PBSA | 18.3 ± 0.5 | 74.1 ± 1.8 | 144.1 ± 0.2 | ||
| PLA-CA-PBSA | 24.4 ± 0.5 | 84.9 ± 0.2 | 127.0 ± 0.2 | 141.6 ± 0.2 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Xu, H.; Odelius, K.; Hakkarainen, M. Poly(lactide)-g-poly(butylene succinate-co-adipate) with High Crystallization Capacity and Migration Resistance. Materials 2016, 9, 313. https://doi.org/10.3390/ma9050313
Yang X, Xu H, Odelius K, Hakkarainen M. Poly(lactide)-g-poly(butylene succinate-co-adipate) with High Crystallization Capacity and Migration Resistance. Materials. 2016; 9(5):313. https://doi.org/10.3390/ma9050313
Chicago/Turabian StyleYang, Xi, Huan Xu, Karin Odelius, and Minna Hakkarainen. 2016. "Poly(lactide)-g-poly(butylene succinate-co-adipate) with High Crystallization Capacity and Migration Resistance" Materials 9, no. 5: 313. https://doi.org/10.3390/ma9050313