
Transient Biocompatible Polymeric Platforms for Long-Term Controlled Release of Therapeutic Proteins and Vaccines


 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
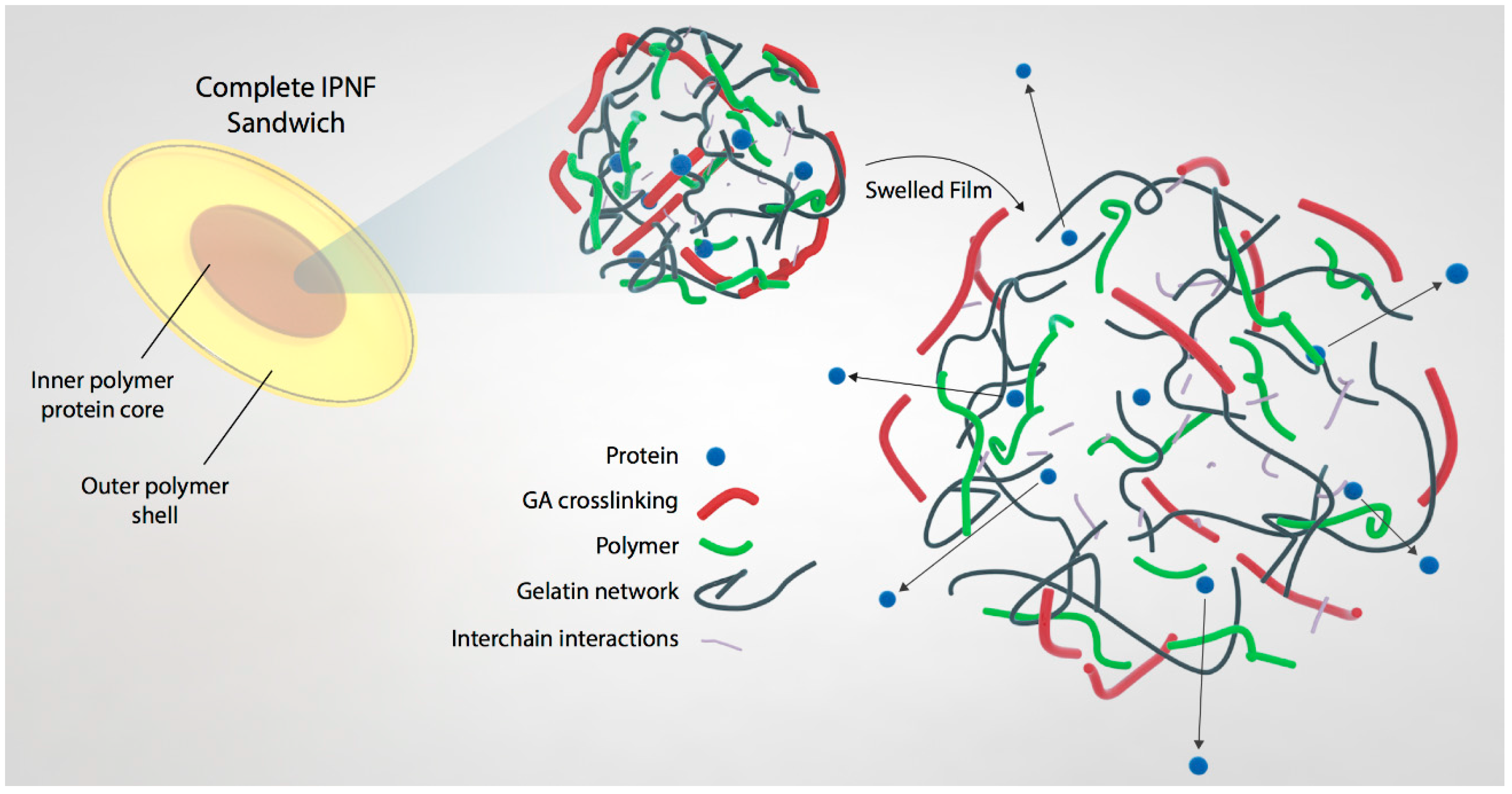
2.2. IPNF Preparation
2.3. Gravimetrical Analysis of Degradation
2.4. Cytotoxicity Evaluation
2.5. Preparation of Protein Loaded Pellets
2.6. In Vitro Release of Protein
2.7. ELISA Test
3. Results and Discussion
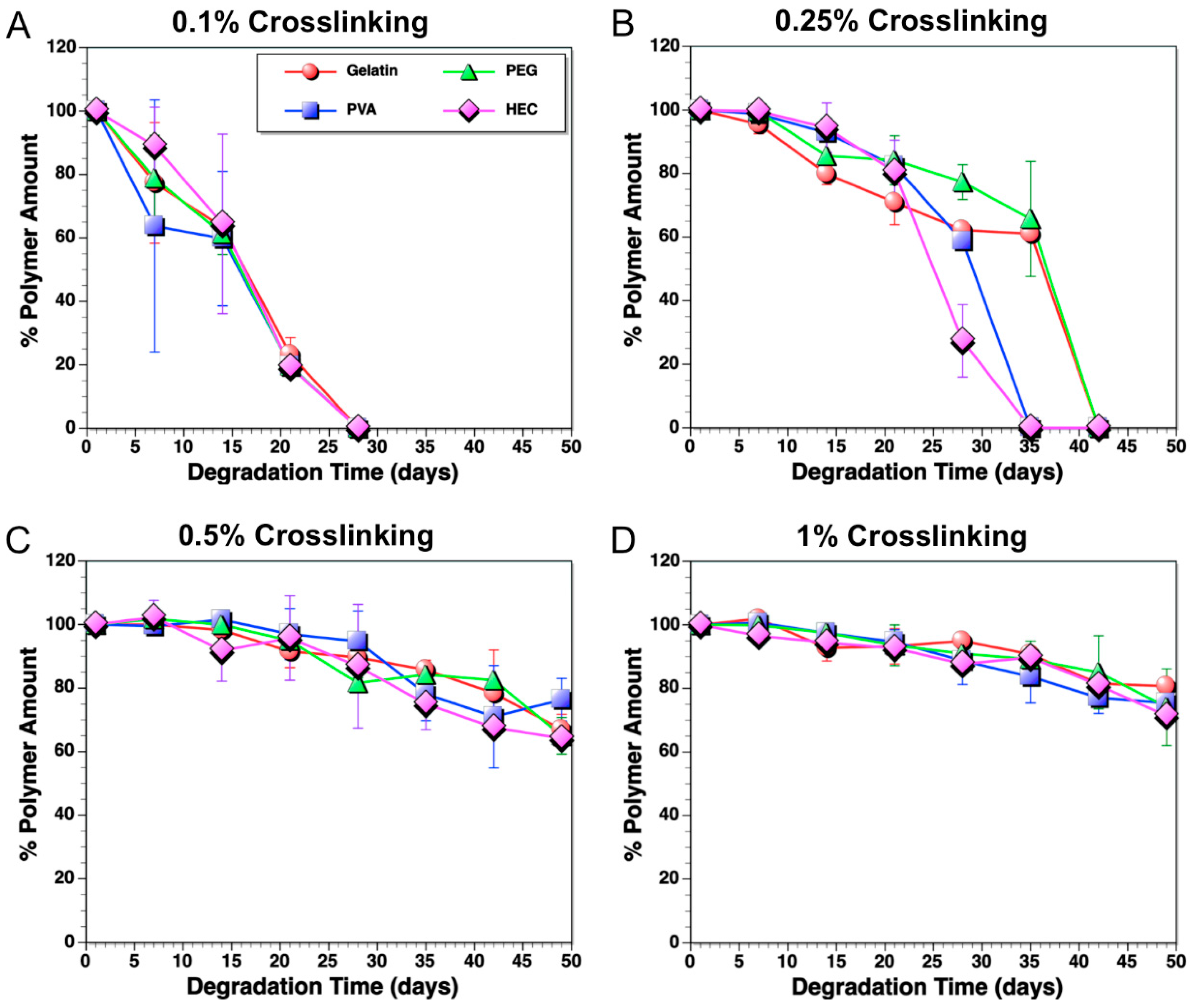
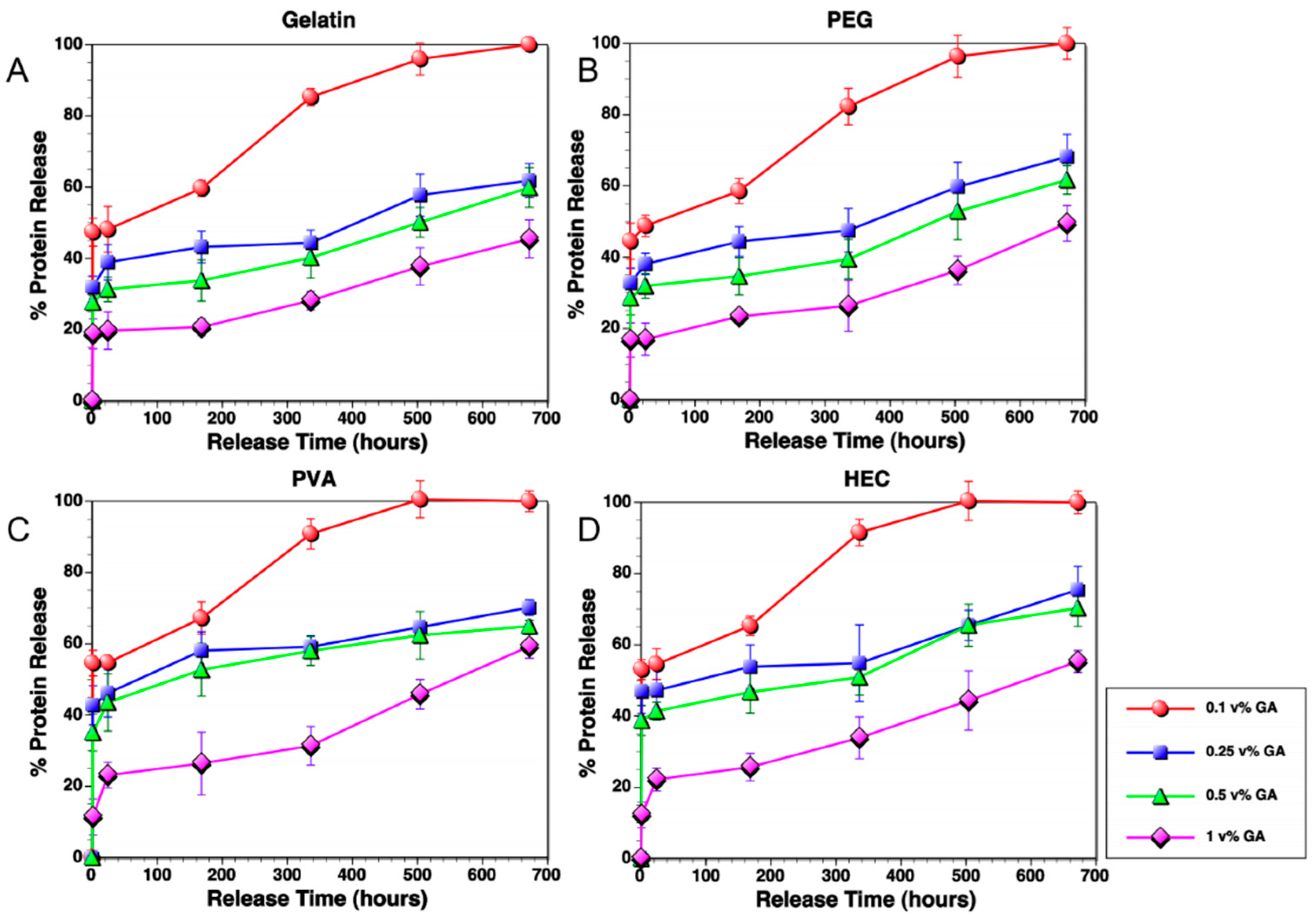
3.1. Degradation Profiles of Polymer Composites
3.2. Cytotoxicity Assay
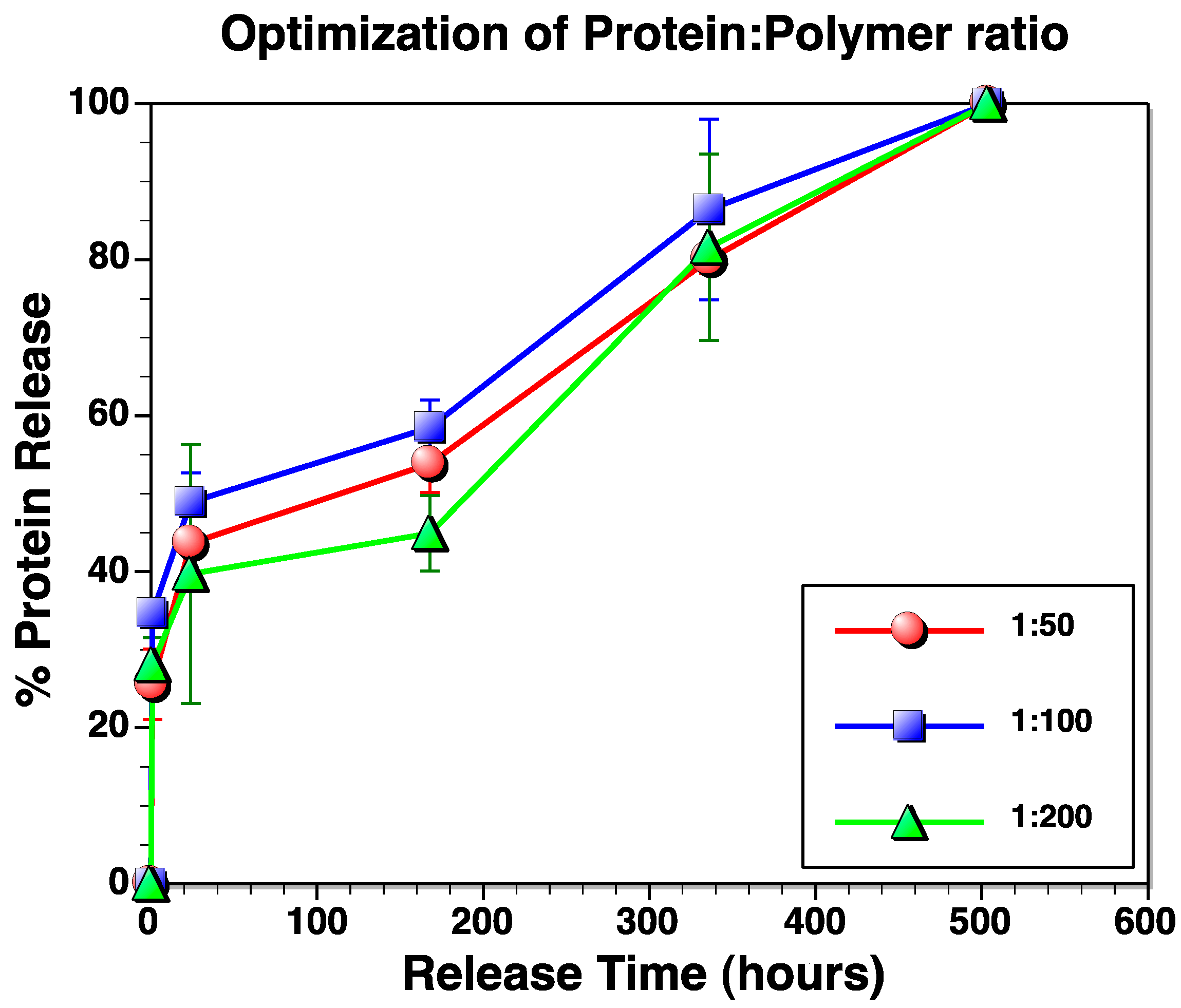
3.3. Determination of Protein Loading Capacity of IPNFs
3.4. Protein Release Kinetics
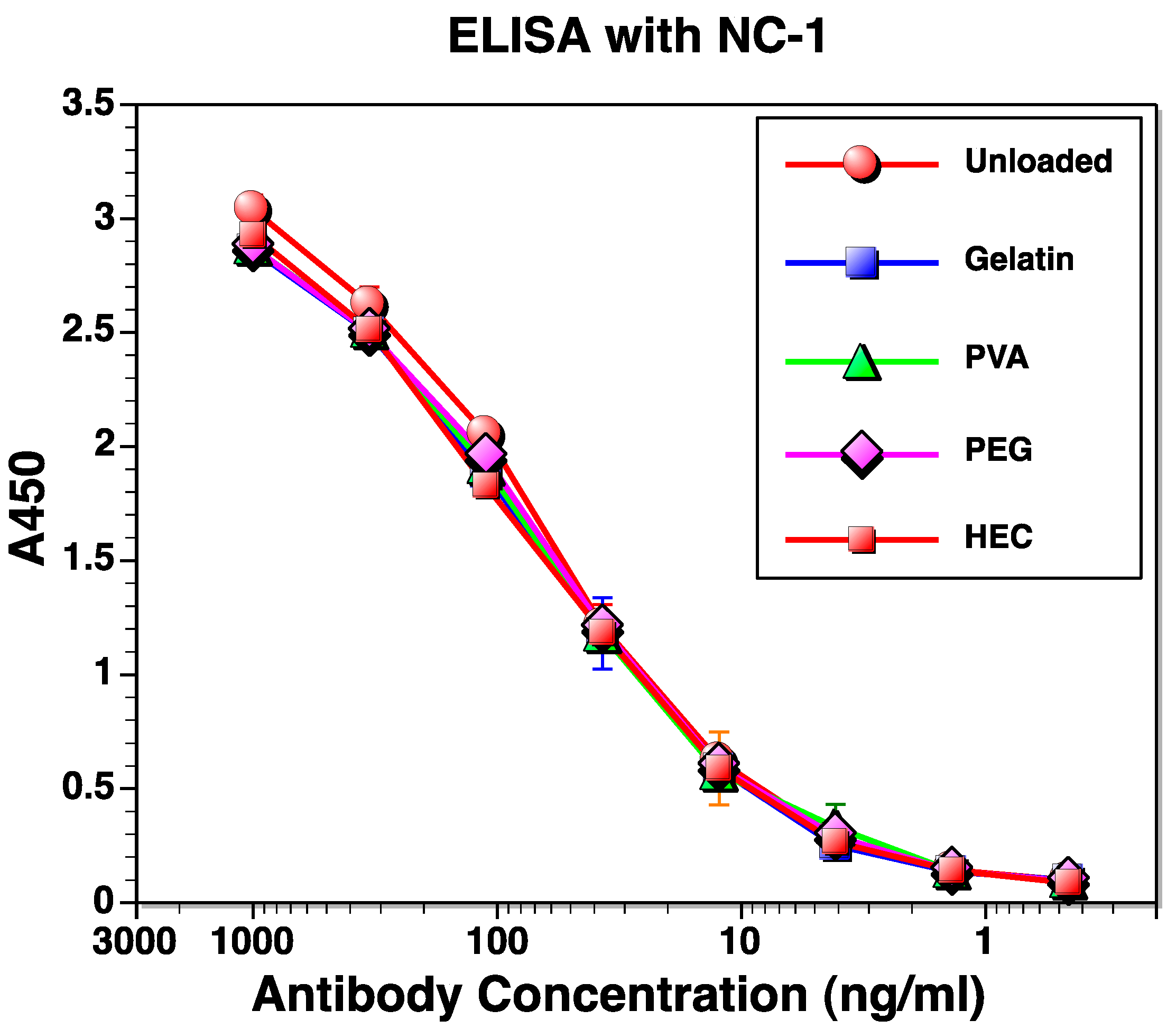
3.5. Determination of Protein Structure upon Release
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, S.; Montazami, R.; Liu, Y.; Jain, V.; Lin, M.; Heflin, J.R.; Zhang, Q.M. Layer-by-layer self-assembled conductor network composites in ionic polymer metal composite actuators with high strain response. Appl. Phys. Lett. 2009, 95. [Google Scholar] [CrossRef]
- Liu, S.; Montazami, R.; Liu, Y.; Jain, V.; Lin, M.; Zhou, X.; Heflin, J.R.; Zhang, Q.M. Influence of the conductor network composites on the electromechanical performance of ionic polymer conductor network composite actuators. Sens. Actuators A Phys. 2010, 157, 267–275. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, S.; Lin, J.; Wang, D.; Jain, V.; Montazami, R.; Heflin, J.R.; Li, J.; Madsen, L.; Zhang, Q.M. Ion transport and storage of ionic liquids in ionic polymer conductor network composites. Appl. Phys. Lett. 2010, 96. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, R.; Ghaffari, M.; Lin, J.; Liu, S.; Cebeci, H.; de Villoria, R.G.; Montazami, R.; Wang, D.; Wardle, B.L.; et al. Equivalent circuit modeling of ionomer and ionic polymer conductive network composite actuators containing ionic liquids. Sens. Actuators A Phys. 2012, 181, 70–76. [Google Scholar] [CrossRef]
- Meis, C.; Hashemi, N.; Montazami, R. Investigation of spray-coated silver-microparticle electrodes for ionic electroactive polymer actuators. J. Appl. Phys. 2014, 115. [Google Scholar] [CrossRef]
- Montazami, R.; Liu, S.; Liu, Y.; Wang, D.; Zhang, Q.; Heflin, J.R. Thickness dependence of curvature, strain, and response time in ionic electroactive polymer actuators fabricated via layer-by-layer assembly. J. Appl. Phys. 2011, 109. [Google Scholar] [CrossRef]
- Montazami, R.; Wang, D.; Heflin, J.R. Influence of conductive network composite structure on the electromechanical performance of ionic electroactive polymer actuators. Int. J. Smart Nano Mater. 2012, 3, 204–213. [Google Scholar] [CrossRef]
- Tsai, H.K.A.; Moschou, E.A.; Daunert, S.; Madou, M.; Kulinsky, L. Integrating biosensors and drug delivery: A step closer toward scalable responsive drug-delivery systems. Adv. Mater. 2009, 21, 656–660. [Google Scholar] [CrossRef]
- Schmidt, D.J.; Moskowitz, J.S.; Hammond, P.T. Electrically triggered release of a small molecule drug from a polyelectrolyte multilayer coating. Chem. Mater. 2010, 22, 6416–6425. [Google Scholar] [CrossRef] [PubMed]
- Bubnova, O.; Khan, Z.U.; Wang, H.; Braun, S.; Evans, D.R.; Fabretto, M.; Hojati-Talemi, P.; Dagnelund, D.; Arlin, J.-B.; Geerts, Y.H.; et al. Semi-metallic polymers. Nat. Mater. 2014, 13. [Google Scholar] [CrossRef]
- Giusti, P.; Lazzeri, L.; de Petris, S.; Palla, M.; Cascone, M.G. Collagen-based new bioartificial polymeric materials. Biomaterials 1994, 15, 1229–1233. [Google Scholar] [CrossRef]
- Hsieh, T.-T.; Hsieh, K.-H.; Simon, G.P.; Tiu, C. Interpenetrating polymer networks of 2-hydroxyethyl methacrylate terminated polyurethanes and polyurethanes. Polymer 1999, 40, 3153–3163. [Google Scholar] [CrossRef]
- Kosmala, J.D.; Henthorn, D.B.; Brannon-Peppas, L. Preparation of interpenetrating networks of gelatin and dextran as degradable biomaterials. Biomaterials 2000, 21, 2019–2023. [Google Scholar] [CrossRef]
- Changez, M.; Burugapalli, K.; Koul, V.; Choudhary, V. The effect of composition of poly(acrylic acid)–gelatin hydrogel on gentamicin sulphate release: In vitro. Biomaterials 2003, 24, 527–536. [Google Scholar] [CrossRef]
- Rivnay, J.; Owens, R.M.; Malliaras, G.G. The Rise of organic bioelectronics. Chem. Mater. 2014, 26, 679–685. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, Q.; Du, Y. Alginate/gelatin blend films and their properties for drug controlled release. J. Membr. Sci. 2006, 280, 37–44. [Google Scholar] [CrossRef]
- Bergo, P.; Moraes, I.; Sobral, P. Effects of plasticizer concentration and type on moisture content in gelatin films. Food Hydrocoll. 2013, 32, 412–415. [Google Scholar] [CrossRef]
- Dhawale, D.S.; Mane, G.P.; Joseph, S.; Anand, C.; Ariga, K.; Vinu, A. Enhanced supercapacitor performance of N-doped mesoporous carbons prepared from a gelatin biomolecule. ChemPhysChem 2013, 14, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, R.; Nastruzzi, C.; Davis, S.S. Sugar cross-linked gelatin for controlled release: Microspheres and disks. Biomaterials 1998, 19, 1641–1649. [Google Scholar] [CrossRef]
- Kanmani, P.; Rhim, J.-W. Physical, mechanical and antimicrobial properties of gelatin based active nanocomposite films containing AgNPs and nanoclay. Food Hydrocoll. 2014, 35, 644–652. [Google Scholar] [CrossRef]
- Tabata, Y.; Ikada, Y. Protein release from gelatin matrices. Adv. Drug Deliv. Rev. 1998, 31, 287–301. [Google Scholar] [CrossRef]
- Tabata, Y.; Nagano, A.; Ikada, Y. Biodegradation of hydrogel carrier incorporating fibroblast growth factor. Tissue Eng. 1999, 5, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Trehan, A. Biodegradable microspheres for protein delivery. J. Control. Release 2003, 90, 261–280. [Google Scholar] [CrossRef]
- Ulubayram, K.; Cakar, A.N.; Korkusuz, P.; Ertan, C.; Hasirci, N. EGF containing gelatin-based wound dressings. Biomaterials 2001, 22, 1345–1356. [Google Scholar] [CrossRef]
- Liu, Y.; Chan-Park, M.B. Hydrogel based on interpenetrating polymer networks of dextran and gelatin for vascular tissue engineering. Biomaterials 2009, 30, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Niu, L.H.; Lai, K.Q.; Huang, Y.Q. Physicochemical properties of biodegradable Tilapia skin gelatin film and gelatin-polysaccharide based composite films. Adv. Mater. Res. 2014, 941. [Google Scholar] [CrossRef]
- Ponez, L.; Sentanin, F.; Majid, S.; Arof, A.; Pawlicka, A. Ion-conducting membranes based on gelatin and containing LiI/I2 for electrochromic devices. Mol. Cryst. Liquid Cryst. 2012, 554, 239–251. [Google Scholar] [CrossRef]
- Bui, A.; Virgilio, N. Tuning organogel properties by controlling the organic-phase composition. Ind. Eng. Chem. Res. 2013, 52, 14185–14191. [Google Scholar] [CrossRef]
- Jamshidi, R.; Çinar, S.; Chen, Y.; Hashemi, N.; Montazami, R. Transient bioelectronics: Electronic properties of silver microparticle-based circuits on polymeric substrates subjected to mechanical load. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 1603–1610. [Google Scholar] [CrossRef]
- Caplin, J.D.; Granados, N.G.; James, M.R.; Montazami, R.; Hashemi, N. Microfluidic Organ-on-a-chip technology for advancement of drug development and toxicology. Adv. Healthc. Mater. 2015, 4, 1426–1450. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, J.E.; Ferry, J.D. Studies of the cross-linking process in gelatin gels. III. Dependence of melting point on concentration and molecular weight. J. Phys. Chem. 1954, 58, 992–995. [Google Scholar] [CrossRef]
- Kuijpers, A.J.; Engbers, G.H.M.; Krijgsveld, J.; Zaat, S.A.J.; Dankert, J.; Feijen, J. Cross-linking and characterisation of gelatin matrices for biomedical applications. J. Biomater. Sci. Polym. Ed. 2000, 11, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, M.; Wang, L.; Qiu, H. Preparation and characterization of chitosan-gelatin/glutaraldehyde scaffolds. J. Macromol. Sci. Part B 2014, 53. [Google Scholar] [CrossRef]
- Khan, S.; Ranjha, N. Effect of degree of cross-linking on swelling and on drug release of low viscous chitosan/poly(vinyl alcohol) hydrogels. Polym. Bull. 2014, 71, 2133–2158. [Google Scholar] [CrossRef]
- Martinez, A.W.; Caves, J.M.; Ravi, S.; Li, W.; Chaikof, E.L. Effects of crosslinking on the mechanical properties, drug release and cytocompatibility of protein polymers. Acta Biomater. 2014, 10, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Panzavolta, S.; Rubini, K. Relationship between triple-helix content and mechanical properties of gelatin films. Biomaterials 2004, 25, 5675–5680. [Google Scholar] [CrossRef] [PubMed]
- Hago, E.-E.; Li, X. Interpenetrating polymer network hydrogels based on gelatin and PVA by biocompatible approaches: Synthesis and characterization. Adv. Mater. Sci. Eng. 2013, 2013. [Google Scholar] [CrossRef]
- Acar, H.; Çınar, S.; Thunga, M.; Kessler, M.R.; Hashemi, N.; Montazami, R. Study of physically transient insulating materials as a potential platform for transient electronics and bioelectronics. Adv. Funct. Mater. 2014, 24, 4135–4143. [Google Scholar] [CrossRef]
- Frey, G.; Chen, J.; Rits-Volloch, S.; Freeman, M.M.; Zolla-Pazner, S.; Chen, B. Distinct conformational states of HIV-1 gp41 are recognized by neutralizing and non-neutralizing antibodies. Nat. Struct. Mol. Biol. 2010, 17, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Naftalin, R.J.; Symons, M.C.R. The mechanism of sugar-dependent stabilisation of gelatin gels. Biochim. Biophys. Acta (BBA) Biomembr. 1974, 352, 173–178. [Google Scholar] [CrossRef]
- Cuq, B.; Gontard, N.; Cuq, J.-L.; Guilbert, S. Selected functional properties of fish myofibrillar protein-based films as affected by hydrophilic plasticizers. J. Agric. Food Chem. 1997, 45, 622–626. [Google Scholar] [CrossRef]
- Thomazine, M.; Carvalho, R.A.; Sobral, P.J.A. Physical properties of gelatin films plasticized by blends of glycerol and sorbitol. J. Food Sci. 2005, 70, E172–E176. [Google Scholar] [CrossRef]
- Tanaka, M.; Iwata, K.; Sanguandeekul, R.; Handa, A.; Ishizaki, S. Influence of plasticizers on the properties of edible films prepared from fish water-soluble proteins. Fish. Sci. 2001, 67, 346–351. [Google Scholar] [CrossRef]
- Krogars, K.; Heinämäki, J.; Karjalainen, M.; Niskanen, A.; Leskelä, M.; Yliruusi, J. Enhanced stability of rubbery amylose-rich maize starch films plasticized with a combination of sorbitol and glycerol. Int. J. Pharm. 2003, 251, 205–208. [Google Scholar] [CrossRef]
- Cherian, G.; Gennadios, A.; Weller, C.L.; Chinachoti, P. Thermomechanical behavior of wheat gluten films: Effect of sucrose, glycerin, and sorbitol. Cereal Chem. 1995, 72, 1–6. [Google Scholar]
- Miller, E.J.; Oldinski, R.A. Poly (vinyl alcohol)-gelatin interpenetrating network hydrogels for tissue engineering applications. In Proceedings of the 2014 40th AnnualNortheast Bioengineering Conference (NEBEC), Boston, MA, USA, 25–27 April 2014.
- Daniele, M.A.; Adams, A.A.; Naciri, J.; North, S.H.; Ligler, F.S. Interpenetrating networks based on gelatin methacrylamide and PEG formed using concurrent thiol click chemistries for hydrogel tissue engineering scaffolds. Biomaterials 2014, 35, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Aminabhavi, T.M.; Nadagouda, M.N.; More, U.A.; Joshi, S.D.; Kulkarni, V.H.; Noolvi, M.N.; Kulkarni, P.V. Controlled release of therapeutics using interpenetrating polymeric networks. Expert Opin. Drug Deliv. 2015, 12, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Banasik, M.; Kim, S.; Penn-Nicholson, A.; Habte, H.H.; LaBranche, C.; Montefiori, D.C.; Wang, C.; Cho, M.W. Eliciting neutralizing antibodies with gp120 outer domain constructs based on M-group consensus sequence. Virology 2014, 462–463, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Rokhade, A.P.; Agnihotri, S.A.; Patil, S.A.; Mallikarjuna, N.N.; Kulkarni, P.V.; Aminabhavi, T.M. Semi-interpenetrating polymer network microspheres of gelatin and sodium carboxymethyl cellulose for controlled release of ketorolac tromethamine. Carbohydr. Polym. 2006, 65, 243–252. [Google Scholar] [CrossRef]
- Wang, Y.; Bansal, V.; Zelikin, A.N.; Caruso, F. Templated synthesis of single-component polymer capsules and their application in drug delivery. Nano Lett. 2008, 8, 1741–1745. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Protein-based nanocarriers as promising drug and gene delivery systems. J. Control. Release 2012, 161, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, B. Mathematical models describing polymer dissolution: Consequences for drug delivery. Adv. Drug Deliv. Rev. 2001, 48, 195–210. [Google Scholar] [CrossRef]
- Shen, E.; Kipper, M.J.; Dziadul, B.; Lim, M.-K.; Narasimhan, B. Mechanistic relationships between polymer microstructure and drug release kinetics in bioerodible polyanhydrides. J. Control. Release 2002, 82, 115–125. [Google Scholar] [CrossRef]
- Speer, D.P.; Chvapil, M.; Eskelson, C.; Ulreich, J. Biological effects of residual glutaraldehyde in glutaraldehyde-tanned collagen biomaterials. J. Biomed. Mater. Res. 1980, 14, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.M.; McRae, G.I.; Vitale, K.M.; Kell, B.A. Controlled delivery of an LHRH analogue from biodegradable injectable microspheres. J. Control. Release 1985, 2, 187–195. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Lu, M. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 1998, 72, 10213–10217. [Google Scholar] [PubMed]
- Attarwala, H.; Amiji, M. Multi-compartmental nanoparticles-in-emulsion formulation for macrophage-specific anti-inflammatory gene delivery. Pharm. Res. 2012, 29, 1637–1649. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Constituents |
|---|---|
| GEL | 5 wt % gelatin |
| GEL-PVA | 5 wt % gelatin + 0.5 wt % PVA |
| GEL-PEG | 5 wt % gelatin + 0.5 wt % PEG |
| GEL-HEC | 5 wt % gelatin + 0.5 wt % HEC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acar, H.; Banerjee, S.; Shi, H.; Jamshidi, R.; Hashemi, N.; Cho, M.W.; Montazami, R. Transient Biocompatible Polymeric Platforms for Long-Term Controlled Release of Therapeutic Proteins and Vaccines. Materials 2016, 9, 321. https://doi.org/10.3390/ma9050321
Acar H, Banerjee S, Shi H, Jamshidi R, Hashemi N, Cho MW, Montazami R. Transient Biocompatible Polymeric Platforms for Long-Term Controlled Release of Therapeutic Proteins and Vaccines. Materials. 2016; 9(5):321. https://doi.org/10.3390/ma9050321
Chicago/Turabian StyleAcar, Handan, Saikat Banerjee, Heliang Shi, Reihaneh Jamshidi, Nastaran Hashemi, Michael W. Cho, and Reza Montazami. 2016. "Transient Biocompatible Polymeric Platforms for Long-Term Controlled Release of Therapeutic Proteins and Vaccines" Materials 9, no. 5: 321. https://doi.org/10.3390/ma9050321
APA StyleAcar, H., Banerjee, S., Shi, H., Jamshidi, R., Hashemi, N., Cho, M. W., & Montazami, R. (2016). Transient Biocompatible Polymeric Platforms for Long-Term Controlled Release of Therapeutic Proteins and Vaccines. Materials, 9(5), 321. https://doi.org/10.3390/ma9050321