PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales)



1
School of life Sciences, Sun Yat-sen University, Guangzhou 510275, China
2
Institute of Ecology, Hunan Academy of Forestry, Changsha 410004, China
3
Hunan Cili Forest Ecosystem State Research Station, Cili 427200, China
*
Author to whom correspondence should be addressed.
Forests 2019, 10(11), 1043; https://doi.org/10.3390/f10111043
Submission received: 9 October 2019
/
Revised: 12 November 2019
/
Accepted: 16 November 2019
/
Published: 18 November 2019
(This article belongs to the Special Issue New Advances in Tree (Epi)Genomics and Implications for Breeding and Management)
Abstract
:The genus Gnetum includes pantropical trees, shrubs and lianas, with unresolved phylogenetic relationships with other seed plant groups. Despite the reference genome for this genus being recently published, the molecular mechanisms that regulate the reproductive organ development of Gnetum remain unclear. A previous study showed that indole-3-acetic acid is involved in the regulation of female strobili of Gnetum, while the diversity and evolution of indole-3-acetic acid-related genes—the Aux/IAA genes—have never been investigated in Gnetales. Thus, a pooled sample from different developmental stages of female strobili in Gnetum luofuense C.Y. Cheng was sequenced using PacBio single-molecular long-read technology (SMRT) sequencing. PacBio SMRT sequencing generated a total of 53,057 full-length transcripts, including 2043 novel genes. Besides this, 10,454 alternative splicing (AS) events were detected with intron retention constituting the largest proportion (46%). Moreover, 1196 lncRNAs were identified, and 8128 genes were found to possess at least one poly (A) site. A total of 3179 regulatory proteins, including 1413 transcription factors (e.g., MADS-box and bHLHs), 477 transcription regulators (e.g., SNF2), and 1289 protein kinases (e.g., RLK/Pelles) were detected, and these protein regulators probably participated in the female strobili development of G. luofuense. In addition, this is the first study of the Aux/IAA genes of the Gnetales, and we identified 6, 7 and 12 Aux/IAA genes from Gnetum luofuense, Welwitschia mirabilis, and Ephedra equistina, respectively. Our phylogenetic analysis reveals that Aux/IAA genes from the gymnosperms tended to cluster and possessed gene structures as diverse as those in angiosperms. Moreover, the Aux/IAA genes of the Gnetales might possess higher molecular evolutionary rates than those in other gymnosperms. The sequencing of the full-length transcriptome paves the way to uncovering molecular mechanisms that regulate reproductive organ development in gymnosperms.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The order Gnetales comprises three extant genera: Ephedra L., Welwitschia Hook.f. and Gnetum L. The gross morphology of the Gnetales, especially their reproductive organs, dramatically differs from that of non-Gnetalean gymnosperms [1,2,3]. The systematic position of the Gnetales remain unresolved among extant seed plant groups, and hypotheses proposed based on morphological and molecular data are constantly contradictory [1,4,5,6,7,8]. Gnetum, comprising around 40 species, is a pantropical genus with the main life-forms of trees, shrubs and lianas [9,10,11]. As dioecious, a female strobilus of Gnetum is featured by several rolls of involucres—for each roll, multiple reproductive units closely connect to a main axis—whereas a male strobilus usually bears one layer of sterile reproductive units and several layers of staminate structures attached by 20–80 microsporangia [1,3,11,12]. Previous studies reveal that the transcription factors of MADS-box genes participate in the development of female and male strobili of Gnetum parvifolium (Warb.) C.Y. Cheng [13] and Gnetum gnemon L. [14,15,16,17]. Moreover, Lan et al. [18] found that the concentration of plant hormones varied across different developmental stages in the female and male strobili of G. parvifolium. Gibberellins A3 and zeatin riboside were regarded as being involved in the sexual identity and subsequent development of male strobili, whereas the increased amount of indole-3-acetic acid promoted the origin and development of female strobili [18].
Plant hormone auxin/indole-3-acetic acid is responsible for plant growth and development, including embryogenesis, lateral root initiation, organogenesis, leaf expansion, inflorescence and fruit development, etc. [19,20]. Aux/IAAs, as transcriptional repression factors, are responsible for the regulation of auxin-response transcription in land plants [21,22,23,24]. Aux/IAAs are characteristic of four conservative domains, designated as domains I–IV, but in certain proteins, one or some domains are lacking [22,23,24]. In addition, Aux/IAAs have two nuclear localization signals (NLS), both of which can connect the nucleus [25,26,27]. Besides this, Aux/IAAs play a role in depressing the activities of other transcriptional regulators, ARFs (ADP-ribosylation factors), through protein–protein interactions [21,22,25,28]. Aux/IAA gene families, to date, have been characterized in several angiosperm species; e.g., 29 genes in Arabidopsis thaliana (L.) Heynh., 31 in Oryza sativa L. [27], and 34 in Zea mays L. [29]. In contrast to angiosperms, knowledge about Aux/IAA gene families in gymnosperms has received less attention, and one study characterized five Aux/IAA; i.e., PTIAA1-5 genes from Pinus taeda [26]. The diversity and functions of Aux/IAA genes in the Gnetales remain poorly understood. A recent study compared the ratio values of nonsynonymous (Ka) and synonymous values (Ks) for paralogous gene pairs of Aux/IAA genes among several seed plant representatives. The species sampling of gymnosperms, however, was limited to P. abies, whose 31 Aux/IAA genes derived from a genome-wide survey [24]. Except for conifers, genes that are involved in the endogenous hormone remain unknown in the Gnetales.
The very recent publication of nuclear genome and Illumina-sequenced transcriptome of varied organs in G. montanum Markgr. [= Gnetum luofuense C.Y. Cheng, 4] provides opportunities to perform a complete survey of the Aux/IAA genes in Gnetum. However, since Illumina-sequencing techniques could not sequence the full length of most transcripts, it is difficult to explicitly elucidate the molecular mechanisms that regulate reproductive organ development in Gnetum. For an example, the alternative splicing (AS) of transcripts remarkably enrich the protein-coding potential in eukaryotic genomes [30,31,32]. Previous studies have shown that ~60% of the multiple exon genes in plants harbor AS transcripts [33]. Besides this, alternative polyadenylation (APA) at the 3′ end of mRNA plays an important role in the transportation, localization and subsequent translation of RNA in most eukaryotic organisms [34,35]. Thus, the alternating cleavage and polyadenylation lead to transcriptome complexity and regulate the gene expression in plants [35,36,37,38]. Due to the short reads generated from Illumina-sequenced data, it is difficult to completely survey AS and APA events [39]. In addition, long noncoding RNAs (lncRNAs), which do not encode proteins, are essential in the gene regulation of eukaryotic cells [40,41,42]. Numerous lncRNAs to date have been identified in several angiosperms; e.g., Arabidopsis thaliana [43], Zea mays [44,45], Medicago truncatula [46] and Populus trichocarpa [47]. In contrast to angiosperms, investigations of lncRNA in gymnosperms are rare, and it is difficult to predict and annotate lncRNAs because of the lack of homologies between closely related species and nonorthologous species [40].
The third-generation sequencing platform, PacBio, is able to generate up to 60-kb-long reads of full-length transcriptomes [39,48]. With single-molecular real-time (SMRT) sequencing, our understanding of AS, APA and lncRNAs is becoming better in angiosperms, as shown in the examples reported in Sorghum bicolour L. Moench [39], bamboo (Phyllostachys edulis (Carrière) J.Houz.) [49], Amborella trichopoda Baill. [50] and strawberry (Fragaria vesca L.) [51], red clover (Trifolium pratense L.) [52], and sugar cane (Saccharum officinarum L.) [53]. In contrast to angiosperms, however, AS, APA and lncRNAs in gymnosperms have never been investigated to date using PacBio sequencing technique. Therefore, in this present study, we adopted the full-length transcriptome from G. luofuense female strobili at different developmental stages using PacBio SMRT sequencing. Illumina-based RNA sequencing was applied to refine the PacBio transcript isoforms by short-read error correction. Further isoform analyses were performed to reveal the diversity of AS isoforms and APA events and lncRNAs. In addition, the diversity of Aux/IAA genes was surveyed in the Gnetales and their phylogeny, structural features, and molecular evolutionary rates were investigated. The first full-length transcriptome of reproductive organs in the Gnetales provides a valuable resource for investigating the reproductive development and evolution of Aux/IAA genes in gymnosperms.
2. Materials and Methods
2.1. Plant Materials and RNA Extraction
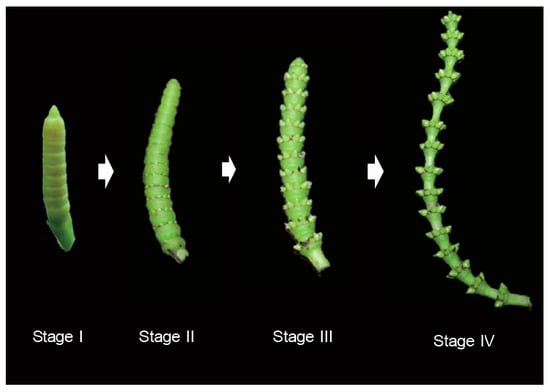
The plant material used for full-length transcriptome sequencing was collected from a mature female individual of G. luofuense cultivated in Bamboo Garden, Sun Yat-sen University on 8 May 2018. To reveal the transcriptome of the entire developmental process, female strobili at four different developmental stages as defined by Lan, Liu, Shi, Deng, Jiang and Chang [18] were pooled (Figure 1), snap-frozen in liquid nitrogen, and stored at −80 °C. The collected material was divided into two samples: one for PacBio SMRT sequencing, and the other for Illumina sequencing. The voucher for the two specimens (No. CH003) was deposited in the herbarium of Sun Yat-sen University, SYS.
Total RNA was extracted from the two samples using an RNA kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. The relic DNA was removed using RNase-free DNase set (Qiagen), and the cleaned RNA samples were assessed by gel electrophoresis with 1% agarose. Besides this, the purity and integrity of extracted RNA was assessed by NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), respectively.
2.2. cDNA Construction and PacBio SMRT Sequencing
The high-quality mRNA (RNA integrity value >7.0) was transcribed into full-length cDNA using a SMARTer PCR cDNA Synthesis kit (Clontech, CA, USA). After PCR amplification, quality control and purification, RNA samples were subjected to cDNA damage/terminal reparation and the connection of SMRT dumbbell-type adapters. A SMRTBell Template library was constructed based on the cDNA production using a SMRTBell Template Prep Kit (Pacific Biosciences, CA, USA). The insert size of the cDNA library was measured by an Agilent 2100 Bioanalyzer, and the concentration of the cDNA library was assessed using Qubit 2.0 flurometer (Life Technologies, CA, USA). One SMRT cell was sequenced on a PacBio RS II platform at Biomarker Technologies Inc., Beijing, China. The PacBio raw reads have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (accession number SAMN12911136).
2.3. Illumina Library Construction and Sequencing
An RNA sample that possesses Poly (A) was segregated from the total RNA using oligo (dT) magnetic beads. A fragmentation buffer was applied to randomly split the enriched RNA into small pieces, and the first strand of cDNA was synthesized using hexamers and reverse transcriptase (Superscript III, Invitrogen). After removing the RNA templates, the second strand of cDNA was subsequently synthesized using deoxyribonucleotide triphosphates (dNTPs), RNase H and DNA polymerase I (Sigma-Aldrich). The synthesized double-strand cDNA was purified with AMPure XP beads. After terminal reparation and A-tailing, the short cDNA fragments were subjected to ligation with Illumina adaptors and purification with AMPure XP beads. The cDNA library was created with PCR amplification, and its quality and concentration were assessed by an Agilent 2100 Bioanalyzer and Qubit 2.0 flurometer, respectively. The final cDNA library was sequenced using an Illumina Hiseq 4000 system. The raw reads generated from Illumina sequencing have been deposited in the NCBI SRA database (accession number SAMN12911137).
2.4. PacBio Long Read Processing and Genome Mapping
Reads of inserts (ROIs) were generated with the following settings of parameters: full passes ≥0, a predicted accuracy of circular consensus sequence (CCS) >0.8 and a minimum length of 50 bp. These ROIs were further divided into full-length (FL) reads and non-full-length (nFL) reads with regard to the presence of 5′ and 3′ cDNA primers and a 3′ poly (A) tail. All ROIs, comprising both FLs and nFLs, were clustered by the Iterative Clustering for Error Correction (ICE) algorithm to obtain consensus isoforms. To obtain high-quality isoforms, full-length nonchimeric (FLNC) transcripts from the Information Concealment Engine (ICE) algorithm were corrected using the Quiver algorithm (https://www.pacb.com/applications/rna-sequencing) with a post-correction accuracy above 99%. The remaining low-quality isoforms were corrected from the Illumina sequenced data with the Proovread tool version 2.12 [54].
Full-length nonchimeric and corrected low-quality transcripts were mapped against the reference genome of G. montanum [= G. luofuense, 4] using GMAP [55] with the following setting parameters: -cross-species, -allow-close-indels=0. The software cDNA Cupcake (https://github.com/Magdoll/cDNA_Cupcake/wik) was applied to remove the redundant transcripts with a minimum alignment accuracy of 0.9 and minimum coverage of 0.85. Mapped transcripts with length differences merely on their 5′ ends were not considered to be redundant. The assessment of transcriptome completeness was performed with regard to the conserved content of transcripts using BUSCO (Benchmarking Universal Single Copy Orthologs) [56]. The mapped transcripts were compared to conserved proteins, and the percentages of aligned and partially aligned transcripts were calculated. In addition, the novel genes were identified, annotated and classified into gene families using Pfam.
2.5. AS and APA Analysis
The GMAP output files in the BAM and gff format were used for subsequent gene structure analysis. Alternative splicing (AS) events were identified using Astalavista (version 3.0, Barcelona, Spain) [57], and five types of AS were identified: alternative 3′ splice site (A3SS), alternative 5′ splice site (A5SS), exon skipping (ES), intron retention (IR), and mutually exclusive exon (MEE). In addition, alternative polyadenylation (APA) was analyzed using the TAPIS pipeline (Colorado, USA) [39]. The detection of motifs was performed on the sequence of 50 nucleotides upstream of poly (A) sites in all transcripts using the web service MEME [58].
2.6. Functional Annotation and Classification
The putative gene function of all corrected isoforms was determined by searching against the following datasets: NR (NCBI nonredundant protein sequences, http//www.ncbi.nlm.nih.gov/), SwissProt (a manually annotated and reviewed protein sequence database, http://www.expasy.org/sprot/), KOG/COG (Clusters of Orthologous Groups of proteins, http://www.ncbi.nlm.nih.gov/COG/), GO (Gene Ontology, http://www.geneontology.org), and KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/) using BLASTX version 2.2.26 with E-value < 1× 10−5. We also searched the database Pfam (Protein Family, http://pfam.xfam.org/) to annotate all the corrected isoforms using HMMER version 3.1b2 [59,60] with E-value < 1× 10−10. Besides this, the KEGG orthology was predicted based on the best-fit BLAST hits from the KEGG database using KOBAS (version 2.0, Beijing, China) [61].
2.7. Identification of CDS, Regulatory Proteins and lncRNAs
The coding sequences (CDS) of corrected isoforms were identified with TransDecoder [62] concerning the length of the open reading frames (ORFs), log-likelihood score and similarity with amino acid sequences in the Pfam database. Based on the putative protein sequences, three types of regulatory proteins—i.e., transcription factors (TFs), transcript regulators (TRs) and protein kinases (PKs)—were identified via searching Plant TFDB4.0 (The Plant Transcription Factor Database 4.0, http://planttfdb.cbi.pku.edu.cn) using iTAK (version 15.03, NY, USA) [63].
To predict lncRNAs (long noncoding RNAs), four analysis methods including CPC (Coding Potential Calculator) [64], CNCI (Coding-Non-Coding Index) [65], CPAT (Coding Potential Assessment Tool) [66], and Pfam were applied to distinguish nonprotein and protein coding transcripts. Putative protein-coding transcripts were filtered out, and the potential lncRNAs fulfilled the thresholds that the length of transcripts should be more than 200 nt and that they should possess at least two exons. The identified lncRNAs were classified into four groups, lincRNA, antisense-lncRNA, sense-lncRNA, and intronic-lncRNA, with regard to the lncRNA location on the reference genome of G. luofuense. In addition, target genes on the genome of G. luofuense regulated by lncRNAs were predicted using LncTar (version 1.0, Tianjin, China) [67]. Two approaches of gene prediction were applied since lncRNAs regulates target genes either by cis or by trans [68,69]. One approach considered that the expression of lncRNAs differs from that of mRNA within every 100 kbp on each chromosome, whereas the other approach merely took the sequence complementation between lncRNAs and mRNAs into account.
2.8. Aux/IAA Gene Identification and Sequences Retrieval
The Aux/IAA genes from G. luofuense were also obtained from the identified TRs of G. luofuense mentioned above and the transcriptome of E. equisetina Bunge [4]. Amino acid sequences of Aux/IAA genes were obtained via the searching of the nuclear genome of G. luofuense [4] and W. mirabilis Hook.f. [70] and Gingko biloba L. [71] using HMMER (version 3.2.1, VA, USA) with an E-value < 1 × 10−5. These putative Aux/IAA genes in the Gnetales and G. biloba were subsequently verified using the NCBI conserved domain database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi?). Moreover, sequences of Aux/IAA genes from other land plant representatives—i.e., A. trichopoda Baill., Picea abies (L.) Karst., and Selaginella moellendorffii Hieron.—which had been verified by by Wu et al. [24], were downloaded from the Phytozome website (http://www.phytozome.org) and PlantGenIE (http://www.plantgenie.org). The sequences of Aux/IAA genes from Arabidopsis thaliana (L.) Heynh. [72], Oryza sativa L. [27], Pinus taeda L. [26] and Zea mays L. [26] were obtained from the GenBank (http://www.ncbi.nlm.nih.gov).
2.9. qRT-PCR Validation and Ka/Ks Analysis
To validate the expression of Aux/IAA genes, six Aux/IAA genes (i.e., GluIAA1-6) were designed using Primer Premier (version 5.0, PREMIER Biosoft International, Palo Alto, CA, USA). Primer sequences and the qRT-PCR protocol are listed in Table S1. Two micrograms of RNA were extracted from the plant material used for PacBio and Illumina sequencing and was subjected to cDNA synthesis. The RT-PCR was performed under the following conditions: 5 min at 95 ℃ (1 cycle), 10 s at 95 ℃, 10 s at 60–65 ℃ and 10 s at 72 ℃ (30 cycles), and 5 min at 72 ℃ (1 cycle). Given the ACTIN gene expression as the reference, the relative expression of six Aux/IAA genes as normalized with the ΔΔCt-method [73].
Sequences of Aux/IAA genes for each species mentioned above were aligned using MUSCLE (version 3.8.31, Cambridge, UK) [74].Values of the nonsynonymous substitution rate (Ka) and the synonymous substitution (Ks) between paralogous gene pairs were calculated using DNAsp (version 6.12.03, Barcelona, Spain) [75] with the default setting. The ratios of Ka/Ks for each gene pair and their standard deviations were calculated and presented in a box plot using ggplot2 package (version 2.2.1, TX, USA) [75] implemented in R version 3.4.4 [76]. The Student t-test—with double tales and different standard deviation—was performed to examine the Ka/Ks values in pairs between different seed plant species. In addition, a phylogeny that comprises 11 land plant species was taken from TimeTree (PA, USA) [77].
2.10. Motif Prediction and Phylogenetic Analysis
A phylogeny was constructed based on 72 full-length Aux/IAA amino acid sequences derived from A. trichopoda, A. thaliana, E. equisetina, G. biloba, G. luofuense, P. taeda, W. mirabilis Hook.f., and Z. mays and was applied to identify motifs using the web server MEME. Three parameters were set according to Wu, Liu, Wang, Li, Liu, Tan, He, Bai and Ma [24]: an optimum motif width of 6–50, a maximum number of motifs of 5, and zero or one occurrence per sequence for each motif site.
Since Aux/IAA genes were dramatically variable, we merely applied the four conservative domains to build up a phylogeny, and the 72 canonical sequences were aligned using MUSCLE. The best-fit model for the amino acid alignment—JTT+Γ—was selected according to AICc values (corrected Akaike Information Criterion) using ProtTest (version 3.2, Vigo, Spain) [78]. A maximum likelihood tree with 1000 pseudo-replicates of simulated bootstraps was created using RAxML-HPC2 version 8.2.12 [79] implemented in web service CIPRES Gateway version 3.3 (http://www.phylo.org). One Aux/IAA gene from Physcomitrella patens (Hedw.) Bruch & Schimp was set as the outgroup.
3. Results
3.1. Assembly of Full-Length Transcriptome and Genome Mapping
PacBio sequencing generated 710,392 ROIs with an average length of 1812 bp, and the mean read quality of each insert was 0.944 (Figure S1A). After the deletion of 30,101 short reads (4.2%), the remaining ROIs were classified into 483,475 full-length reads (FLs, 68.1%), comprising 3551 full-length chimeric reads (0.5%) and 480,225 full-length nonchimeric reads (67.6%, FLNC), and 196,816 non-full-length reads (nFLs, 27.7%) (Figure 2A). The FLNC reads were clustered using the ICE algorithm and yielded 254,573 consensus isoforms with a mean length of 1859 bp (Figure 2B). Together with non-full-length sequences, the consensus isoform in each cluster was polished, resulting in 176,391 high quality isoforms (HQs) and 77,034 low quality isoforms (LQs). All polished consensus isoforms were corrected by Illumina sequencing data and yielded 253,273 corrected isoforms, with an N50 length of 2076 bp and a mean read length of 1837 bp. All corrected isoforms (253, 273) were further mapped against the reference genome with GMAP. After deleting the unmapped and redundant isoforms (18,521, 7.32%), the remaining 234,752 isoforms (92.68%) were assembled into 53,057 transcripts with a mean length of 1992 bp (Figure 2C). The completeness of our transcript dataset was assessed with BUSCO, and the result revealed that this dataset consisted of 61.5% complete and 3.8% partial BUSCO orthologues (Figure 2D).
3.2. Functional Annotation and Novel Gene Detection
The assembled 53,057 transcripts were functionally annotated by searching against the databases NR, SwissProt, KOG, COG, GO, Pfam and KEGG, and a total of 26,305 transcripts (49.6%) were annotated (Figure 3A, Table S2). A total of 25,837 transcripts was annotated by the NR database, and we compared the transcript sequences to the NR databases and found five species (Figure 3B)—i.e., Picea sitchensis (Bong.) Carrière, 7119; A. trichopoda, 2070; Nelumbo nucifera Gaertn., 1729; Vitis vinifera L., 829; and Elaeis guineensis Jacq., 787—obtained the largest five-number of transcripts homologous to G. luofuense. A total of 20,762 transcripts was annotated by Pfam and further classified into 3919 gene families (Table S3). The top five gene families with the largest enriched transcripts were the protein kinase domain (931), protein tyrosine kinase (931), leucine rich repeats (824), pentatricopeptide repeat (PPR) repeat (607), and pentatricopeptide repeat domain (516). The results of GO analysis show that 14,044 transcripts were classified into three categories: “biological process”, “cellular components” and “molecular functions” (Figure 3C). The biological process category was mainly distributed in “metabolic processes” (9409), “cellular process” (6944), and “single-organism process” (5988). Transcripts involved in the cellular component category consisted of “cell part” (6301), “cell” (6133) and “organelle” (4816). Transcripts in the molecular function category were mainly enriched for “catalytic activity” (7806), “binding” (5482), and “transporter activity” (816). Moreover, 10,140 transcripts were annotated by KEGG pathways (Figure 3D), and the largest numbers of the transcripts were enriched in five KEGG pathways: “phenylpropanoid biosynthesis” (387 transcripts, KEGG orthology number, ko00940), “starch and sucrose metabolism” (351, ko00500), “ribosome” (319, ko03010), “protein processing in endoplasmic reticulum” (314, ko04141), and “spliceosome” (300, ko03040). In addition, 2043 novel genes were detected, and all novel gene sequences were listed in Appendix A1. Of the identified novel genes, 1295 genes (63.38%) were annotated by searching against the following databases: NR, SwissProt, KOG, COG, GO, Pfam and KEGG (Figure 3A). A total of 1254 transcripts was annotated by the NR database, and these transcripts were found to be mainly homologous to the five species (Figure 3B): Picea sitchensis, 291; Amborella trichopoda, 40; Nelumbo nucifera, 38; Vitis vinifera, 56; and Elaeis guineensis. 31. GO analysis showed that 532 transcripts were classified into three categories—“biological process”, “cellular components” and “molecular functions” (Figure 3C)—and 247 transcripts were mapped to 25 KEGG pathways (Figure 3D).
3.3. AS and APA Analysis
After aligning to the reference genome, a total of 10,454 AS events was detected, including 4745 intron retention (46%), 2531 alternative 3′ splice sites (24%), 1800 exon skippings (17%), 1235 alternative 5′ splice sites (12%), and 143 mutually exclusive exons (1%) (Figure 4A, Table S4). In total, 9155 genes were detected to possess alternative splicing events, with the isoform numbers ranging one to 55. The largest isoform number—55—was found among three genes: TnS000130997g02, TnS000368105g01, and TnS000008647g03.
To identify the differential polyadenylation sites at the 3′ end of transcripts, a total of 8128 genes was found to possess at least one poly (A) site, of which 1405 genes possess at least five poly (A) sites (Figure 4B, Table S5). The nucleotide composition both upstream (50 nts) and downstream (50 nts) was analyzed to assess the nucleotide bias of all poly (A) cleavage sites. We found an enrichment of uracil (U) 50 nts upstream and adenine (A) downstream of the cleavages site in 3′ UTRs (untranslated regions, Figure 4C). Moreover, we performed a MEME analysis to detect motifs at 50 nts upstream of the poly (A) site of all transcripts to identify cis-elements for polyadenylation. Three conserved motifs—(AAAACA), (GGGCGC) and (CGCCGC)—were identified 25 nts upstream of poly (A) sites (Figure 4D).
3.4. Identification of CDS, Regulatory Proteins and lncRNAs
To identify CDS, 31,928 ORFs were identified with a mean length of 2076 bp with Transdecoder. We found 29,430 5′ UTRs with a mean length of 466 bp and 31,714 3′ UTRs with a mean length of 577 bp. Of the detected ORFs, 24,605 (77%) with a mean length of 2174 bp were CDS, which were composed of start and stop codons. The number and length distribution of the identified CDS are displayed in Figure 5A. Based on the CDS detected, a total of 3179 regulatory proteins was identified, including 1413 transcription factors (TFs), 477 transcript regulators (TRs) and 1289 protein kinases (PKs) (Figure 5B). The amino acid sequences of the identified regulatory proteins are listed in Appendix A2.
In addition, a total of 1196 lncRNAs was identified by the four methods—CNCI, PFAM, CPAT and CPC—with a mean length of 1258 nts (Figure 5C). All sequences of the identified lncRNA are listed in Appendix A3. The detected lncRNAs are classified as lincRNA (488), antisense lncRNAs (20), intronic lncRNA (238), sense lncRNAs (417) and others (33) (Figure 5D). Besides this, 1196 lncRNAs were predicted to regulate considerably different gene numbers using two prediction approaches: 628 genes were regulated by 722 lncRNAs in cis (Table S6), while 11,967 genes were regulated by 1195 lncRNAs in trans (Table S7).
3.5. Aux/IAA Gene Identification, qRT-PCR and Adaptive Evolution Analysis
This is the first study investigating Aux/IAA genes in the Gnetales, and a total of six Aux/IAA genes were found among the transcription regulators (TRs) in G. luofuense: TnS000498063g52 (named GluIAA1 hereafter), TnS000653177g04 (GluIAA2), TnS000867017g28 (GluIAA3), TnS000053353g02 (GluIAA4), TnS000142615g19 (GluIAA5), TnS000994775g01 (GluIAA6). This number of Aux/IAA genes is congruent with that derived from a genome-wide search of G. luofuense. The expression of the six Aux/IAA genes identified was further verified with qRT-PCR (Figure 6A). In addition, we found seven Aux/IAA genes in W. mirabilis, 12 genes in E. equisetina, and six genes in G. biloba. All amino acid sequences of the identified Aux/IAA genes are listed in Appendix A4.
A pairwise comparison of Ka/Ks values among Aux/IAA genes reveals that the average Ka/Ks values in G. luofuense and E. equisetina were both above 1, whereas the average Ka/Ks value was 1 in W. mirabilis (Figure 6B). There was no significance between every two gnetalean genera with regard to Ka/Ks values in pairs using Student t-test: however, the Ka/Ks values of G. luofuense were significantly larger than those in G. biloba (p-value = 5.89 × 10−8) and P. taeda (p-value = 7.73 × 10−4), respectively, whereas no significant difference was found between G. luofuense and P. abies (p-value = 0.2281). Besides, the Ka/Ks values of G. luofuense were significantly larger than those of Z. mays (p-value = 2.18 × 10−3) while significantly smaller than that of A. thaliana (p-value = 1.61 × 10−3) and S. moellendorffii (p-value = 1.82 × 10−6).
3.6. Motif Prediction and Phylogenetic Analysis
The motif composition of Aux/IAA genes was investigated with MEME, and the results show that five conservative motifs were identified (Figure 6C): motif 1, corresponding to domain I of Aux/IAA genes, was characterized by possessing a conservative sequence “LxLxLx”; motif 2, corresponding to domain II, had a conservative sequence “VGWPPV”; motif 3, corresponding to domain III, was characteristic of conservative sequences “VKVxMDG…RK”; motif 4, corresponding to domain IV, had conservative sequences “GDxMLVGDVPW” and “KRxRxMK”; and motif 5 was characterized by possessing newly detected conservative sequences “GLAPRxxEK”. The structures of the detected Aux/IAA gene varied, and a total of eight types—1-2-3-4, 1-2-3-4-5, 1-2-3, 1-5-2-3, 3-4, 2-3-4-5, 2-3-4, and 1-3-4—were identified.
The deep divergence of the phylogeny based on canonical Aux/IAA sequences in different seed plant groups usually has low statistical support (Figure 6B). Nevertheless, the results of phylogenetic analysis reveal that the 72 Aux/IAA genes were divided into two groups: type A and type B genes. Type A genes formed a monophyletic group with good support (clade A, bootstrap values, BS=85) while type B formed two paraphyletic groups—i.e., clades B and C—but with low support. Moreover, Aux/IAA sequences from gymnosperms tend to form a clade, as with the cases found in clades D, E, F, G, H and I, but all the clades received low statistic support.
4. Discussion
4.1. Full-Length Transcriptome and Gene Annotation
A total of 253,273 corrected isoforms was generated in the present study, with a mean read length of 1837 bp. The full-length transcripts can overcome the problems confronted by the short-read sequencing of conventional RNA sequencing. Using PacBio sequencing technique, 31,928 ORFs and 2043 novel genes were identified, suggesting that full-length transcript sequence information provides useful resources to improve genome annotation in G. luofuense. A final of 53,057 transcripts were assembled, and 26,305 genes from 3896 gene families were annotated against the seven databases (NR, SwissProt, KOG, COG, GO, Pfam and KEGG) (Figure 3A). Among the ten KEGG pathways annotated (Figure 3D), “starch and sucrose metabolism”, “purine metabolism”, “ribosome”, “biosynthesis of amino acids”, and “carbon metabolism” might participate in the secretion of sugary pollination drops in G. gnemon and G. luofuense as the response to insect pollination [80]. Besides, KEGG pathways in “phenylpropanoid biosynthesis”, “plant hormone signal transduction”, and “plant–pathogen interaction” might affect fertile reproductive unites development of the two species [80]. Besides, the annotated genes were enriched in the top ten GO enrichment categories (Figure 3C). Our results are congruent with that eight categories i.e., “metabolic process”, “cellular process”, “response to stimulus”, “catalytic activity”, “binding”, “cell part”, “cell” and “organelle” are all densely enriched in the transcripts of female reproductive organs in Gingko [81] and conifers [82]. These results provide useful information to infer the conservative functional genes in regulating the development of female strobili in gymnosperms.
4.2. AS Analysis
In the present study, we found ~46% cases of intron retention, ~24% of three alternative 3′ splice sites, ~17% of exon skipping, ~12% of five alternative 3′ splice sites, and ~1% mutually exclusive exons (Figure 4A). Our results significantly differ from the proportion of AS events identified in Arabidopsis, with ~40% intron retention, ~15.5% three alternative 3′ splice sites, ~17% exon skipping, ~7.5% five alternative 3′ splice sites, and ~8% mutually exclusive exons [31]. A similar proportion of intron retention (~40%) was detected in other angiosperms; e.g., maize [83] and sorghum [39]. In other species of angiosperms, intron retention also accounts for the majority of cases, such as bamboo [49], Amborella trichopoda [50] and strawberry [51], red clover [52], sugar cane [53]. Thus, the results corroborate the suggestion that intron retention constituted the majority of AS events in seed plants [84]. In addition, alternative spliced genes are usually over-represented in order to respond to abiotic stress in angiosperm plants [85,86]. Compared to angiosperms, AS events that associated with the resistance of abiotic stress have been rarely reported in gymnosperms. One study shows that the retention of the first intron in genes that encodes β-hydroxyacyl ACP dehydratase was associated with response to cold temperature during seed imbibition and the regulation of the lipids biosynthesis in Picea mariana [87].Another case shows that anthocyanidin synthase gene in Gingko biloba (named GbANS), which possesses alternative splicing, participate into a series of abiotic stresses, e.g., UV-B, cold, and abscisic acid etc. [88]. Accordingly, the 10,454 AS events with varied types detected in the female strobili of G. luofuense pave the way to detecting the responses of reproductive organs in gymnosperms to abiotic stress in further studies.
4.3. APA Analysis
Besides AS events, alternative polyadenylation is able to produce different lengths of transcripts and contributed to the diversity of mRNA [35,89,90]. APA events affect different methods of 3′ termination, which comprise the protein coding region and 3′ UTRs [35,38,89,91]. A previous study shows that alternative polyadenylation of genes Lhcb2 and Lhcb3, affects the stability and transcription of mRNAs during the leaf development of G. biloba [92]. In angiosperms, the different types of the cleavage and polyadenylation of mRNAs play a role in floral development and the flowering time of Arabidopsis [90,93,94], the detection of APA events in the present study, a total of 8128 genes to have poly A sites (Figure 4B), is helpful for elucidating the female strobili development in G. luofuense. Moreover, we found a clear bias towards nucleotide composition 50 nt upstream and downstream of all poly (A) cleavages (Figure 4C). The nucleotide composition bias found in G. luofuense is congruent with angiosperms (e.g., Arabidopsis [36,37] and Sorghum [39]) and non-Gnetalean gymnosperms (e.g., Gingko [95]). In addition, we found three overrepresented motifs—(AAAACA), (GGGCGC) and (CGCCGC)—at 25 nucleotides upstream from the poly (A) sits of all the transcripts (Figure 4D). These motifs, however, all remarkably differ from a significantly over-represented motif—(AAUAAA) detected in angiosperms (e.g., Arabidopsis [39] and Trifolium [52]) and non-Gnetalean gymnosperms (e.g., Pinus [96] and Gingko [95,97]).
4.4. LncRNA Analysis
Our results show that a total of 1196 lncRNAs with the mean length 1258 bp were identified for the major developmental stages of G. luofuense female strobili. The lncRNAs in G. luofuense were shorter than the full-length transcripts with the mean length of 1992bp, which is consistent with the lncRNAs detected in Picea abies [98]. We found 488 lincRNA (40.80%), 20 antisense-lncRNA (1.67%), 238 intronic-lincRNA (19.90%), 417 sense-lncRNA (34.87%) and 33 others (2.76%). The number and proportion of lncRNAs in G. luofuense dramatically differ from the 1323 lncRNAs from G. biloba, which possessed 762 lincRNA (57.60%), 211 antisense-lncRNA (15.95%), 100 intronic-lincRNA (7.56%) and 280 sense-lncRNA (21.16%) [99]. Besides, a very recent published full-length trancriptome of G. bioloba consisted of 1270 lncRNAs, comprising 628 lincRNA (49.45%), 88 antisense-lncRNA (6.93%), 259 intronic-lincRNA (20.39%), 267 sense-lncRNA (21.02%) and 28 others (2.20%) [95]. The pronounced interspecific and infraspecific variation in lncRNA numbers corroborates the statement that evolutionarily conserved lncRNAs account for quite a small proportion of RNAs, making up 2–5.5%, probably due to the consequence of rapid evolution [100,101,102]. In addition, lncRNAs have effects on their target genes in the manner of either in cis or in trans [99]. Provided that the concentration of lncRNA is at a low level, they tend to execute in cis, regulating the expression of the genes in the vicinity, whereas at a higher level, lncRNAs can perform in trans instead, acting on the genes regardless of their location [68,69]. Our results show that 1093 target genes were predicted to be regulated by lncRNA in cis and 11,967 genes in trans. These results pave the way to characterizing the functions of lncRNAs in the organ development of Gnetum.
4.5. Regulator Proteins
The identification of regulator proteins is important to elucidate the molecular mechanism that regulates the female strobili development of G. luofuense. Our results also show that a total of 3179 regulatory proteins from 176 families were identified, comprising 1413 transcription factors (TFs), 477 transcript regulators (TRs), and 1289 protein kinases (PKs) (Figure 4B). TFs of MADS-box genes play an essential role in reproductive organ evolution of seed plants [17,103]. We found 38 TFs belong to the MADS-box gene family, of which 24 TFs derive from MIKCc-group genes. One previous study reported that three MIKCc-group genes—GpMADS1, GpMADS3 and GpMADS4—were involved in the female strobili development of G. parvifolium [13]. Besides this, MIKCc-group genes—e.g., GGM2, GGM3, GGM9 and GGM11—participate in the sexual determination and development of female strobili in G. gnemon [14,17,104]. A genome-wide research of MADS-box genes yielded a total of 35 MIKCc-group genes [105], the majority of which were detected to express female strobili and fertile reproductive ovules of G. luofuense [80].
TRs of SNF2 genes were shown to participate in the shoot development and flowering in A. thaliana [106]. In the present study, 31 SNF2 TRs in the female strobili of G. luofuense were identified. In addition, a previous study reported a series of membrane-bound transcription factors in seed plants via the whole genome research, e.g., 3 bHLHs, 4 C2H2, and 5 WRKY TFs in Picea abies [107]. We identified considerably more TFs—92 C2H2, 109 bHLH, and 41 WRKY TFs in G. luofuense. The C2H2, bHLH and WRKY TFs were reported to differentially express during floral organ development in Brassica rapa [108]. Besides this, bHLH TFs mediate the carpel margin tissues and petal sizes of Arabidopsis’ flowers [109,110]. Thus, the identified membrane-bound transcription factors might play roles in the female strobili development of G. luofuense. Moreover, we identified 795 RLK/Pelle PKs, accounting for a relatively large proportion (25%) of the detected regulator proteins. The RLK/Pelle family is the largest family of PKs, and over 600 and 1000 RLK/Pelle PKs were found in Arabidopsis and rice, respectively [111]. One gene, CLAVATA1, facilitates the enlargement of the shoot and flower meristems, suggesting an important role in controlling meristem size and floral organ development in A. thaliana [112,113]. In gymnosperm, one gene PsRLK was examined to involve in the seedling development of in Pinus Sylvestris, this gene was strictly expressed in the specialized phloem cells and functioned as a putative receptor-like protein [114].
4.6. Diversity and Molecular Evolutionary Rates of Aux/IAA Genes
Aux/IAA genes are a large gene family that is involved in varied developmental processes of plants including embryogenesis, apical dominance, vascular differentiation, lateral root imitation and shoot elongation [23,24,115]. We found 82 Aux/IAA transcription factors from G. luofuense, corresponding to six genes (namely GluIAA1–GluIAA6), and the expression of the six genes was further validated by qRT-PCR (Figure 6A). Besides of the Aux/IAA genes from Gnetum, we found seven genes from W. mirabilis (i.e., WmiIAA1-WmiIAA7) and 12 genes from E. equistina (i.e., EeqIAA1-EeqIAA12), respectively. The Aux/IAA gene numbers of the three gnetalean genera were all considerably less than those from 13 species (ranging from 16 in A. trichopoda to 63 from G. max) that represent major lineages of angiosperms and P. abies (31) [24]. Considering the low abundance of Aux/IAA genes detected in P. patens (3), S. moellendorffii (9), and G. biloba (6), the diversity of Aux/IAA genes probably dramatically increased during the evolution of land plants. The statement is in accordance with the previous study [24]. In addition, the molecular evolutionary rates were estimated with regard to Ka/Ks values using paralogous Aux/IAA gene pairs for seed plants (Figure 6B). We found the Ka/Ks values of Aux/IAA paralogous gene pairs were overlapping within the Gnetales. It is noticeable that the average Ka/Ks value in G. luofuense and E. equisetina were both above 1, implying a relatively large proportion of the Aux/IAA paralogous gene pairs might experience positive selection during the course of evolution. The average Ka/Ks value of W. mirabilis was close to 1, suggesting the trend of neutral selection for Aux/IAA paralogous gene pairs in W. mirabilis. In contrast, however, a relatively large proportion of Ka/Ks values in P. abies and all Ka/Ks values of G. biloba and P. taeda were lower than 1, implying that these Aux/IAA paralogous gene pairs experienced purifying selection. Thus, Aux/IAA genes might have evolved faster than those in other non-Gnetalean gymnosperms. Compared to gymnosperms, Ka/Ks values of Aux/IAA paralogous gene pairs exhibited a more diverse pattern among the four angiosperm species, which is in accordance with the results found in a previous study [24].
4.7. Phylogenetic Analysis and Structural Detection of Aux/IAA Genes
Phylogenetic analysis is a powerful method to understand the evolution of Aux/IAA genes. Previous studies have made efforts to resolve the phylogenetic relationships of Aux/IAA genes for many angiosperms—e.g., O. sativa [27], Solanum lycopersicum L. [72], and Z. mays [29]—and one gymnosperm species—P. taeda [26]. A recent phylogeny of Aux/IAA genes was constructed based on 253 canonical sequences from 17 land plant species, and was resolved into five clades i.e., clade A-E [24]. The taxon sampling of this 17-species phylogeny; however, this was biased, with merely one gymnosperms species (i.e., P. abies) sampled. A new phylogeny of the present study included the Aux/IAA genes in G. biloba and three Gnetalean genera and shows that the Aux/IAA genes were classified into two types; i.e., type A and type B (Figure 6C). This classification is largely in accordance with that defined in a previous study [27]. Aux/IAA genes from the Gnetales and other gymnosperms tend to segregate from those from angiosperms (examples shown in clades D, E, F, G, H and I, Figure 5C). Nevertheless, the statement requires further validation based on a well-resolved phylogeny of Aux/IAA genes with an extensive sampling of seed plants. In addition, a total of five motifs was identified (Figure 6C), of which four motifs were detected as the four conservative domains of Aux/IAA genes [22,23,24]. Interestingly, a new motif with the conservative amino acids “GLAPRxxEK” was identified, which is located in the vicinity of domain IV. The new motif is commonly seen in gymnosperms, especially in the Gnetales, with quite a few present in angiosperms. Moreover, considering the gene structure, a total of eight types were identified for the 72 Aux/IAA genes from the land plant species. The results corroborate the suggestion that Aux/IAA genes already existed before the divergence of gymnosperms and angiosperms [24,26].
5. Conclusions
This is the first full-length transcriptome of reproductive organs in gymnosperms using PacBio SMRT sequencing. A total of 53,057 transcripts were assembled and 2043 novel genes were identified, improving genome annotation and gene function during the female strobili development of G. luofuense. In addition, AS, APA and lncRNA for all detected transcripts were reported and discussed, paving the way for the analysis of post-transcriptional modification and identification of the regulation mechanism in further studies of reproductive organs in Gnetum. Finally, this is the first study reporting Aux/IAA genes in the Gnetales, and a total of six, seven and 12 Aux/IAA genes were identified for Gnetum, Ephedra and Welwitschia, respectively. The results of phylogenetic analysis and molecular rate assessment corroborate the suggestion that Aux/IAA genes in the Gnetales might have experienced a different evolutionary course from those of other non-Gnetalean groups. Additional work is needed to elucidate the molecular pathways and regulation of the reproductive organs in association with Aux/IAA genes in the Gnetales.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4907/10/11/1043/s1, Figure S1: ROI and FLNC length distribution; Table S1: Primer information of Aux/IAA genes and qRT-PCR protocol; Table S2: All transcripts annotated by searching against varied databases; Table S3: Gene families and transcript information annotated by searching the Pfam database; Table S4: Alternative splicing events and location sites on the reference genome of G. luofuense; Table S5: Information of polyadenylation events detected from G. luofuense female strobili; Table S6: lncRNAs and corresponding cis-regulated genes; Table S7: LncRNAs and corresponding cis-regulated genes.
Author Contributions
Conceptualization, C.H. and N.D.; Funding acquisition, Y.S.; Investigation, C.H. and N.D.; Project administration and supervision, Y.S.; Writing—original draft, C.H.; Writing—review & editing, C.H. and Y.S. All authors contributed to the drafts and gave final approval for publication.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31670200 and 31872670) and the Natural Science Foundation of Guangdong Province, China (No. 2017A030313122 to Y.S. and No. 2018A0303130163 to C.H.).
Acknowledgments
We thank Demei Wang and Guanshen Liu (Biomarker Technologies, Inc.) for providing valuable technical and analytical assistance. We thank Tao Wan, Zhiming Liu, Min Yang (Fairy Lake Botanical Garden, Shenzhen & Chinese Academy of Science) and Rongkai Wang (Bioeditas Technology Corporation-Shaanxi, China) for providing data resources and valuable technical assistance. We also thank two reviewers for valuable comments on the text.
Conflicts of Interest
The authors declare no conflicts of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Appendix A
Appendix A provides all sequence information in the present study: Appendix A1: All sequences of novel genes; Appendix A2: All sequences of regulator proteins; Appendix A3: All lncRNAs detected; Appendix A4: Aux-IAA genes detected from Gnetales and Gingko biloba.
References
- Ickert-Bond, S.M.; Renner, S.S. The Gnetales: Recent insights on their morphology, reproductive biology, chromosome numbers, biogeography, and divergence times. J. Syst. Evol. 2015, 54, 1–16. [Google Scholar] [CrossRef]
- Kubitzki, K. General Traits of the Gnetales. In The Families and Genera of Vascular Plants; Kramer, K.U., Green, P.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 378–379. [Google Scholar]
- Endress, P.K. Structure and function of female and bisexual organ complexes in Gnetales. Int. J. Plant Sci. 1996, 157, 113–125. [Google Scholar] [CrossRef]
- Wan, T.; Liu, Z.M.; Li, L.F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.J.; Xin, H.P.; Gong, Y.B.; Liu, Y.; et al. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82–89. [Google Scholar] [CrossRef]
- Bowe, L.M.; Coat, G.; dePamphilis, C.W. Phylogeny of seed plants based on all three genomic compartments: Extant gymnosperms are monophyletic and Gnetales’ closest relatives are conifers. Proc. Natl. Acad. Sci. USA 2000, 97, 4092–4097. [Google Scholar] [CrossRef]
- Donoghue, M.J.; Doyle, J.A. Seed plant phylogeny: Demise of the anthophyte hypothesis? Curr. Biol. 2000, 10, 106–109. [Google Scholar] [CrossRef]
- Doyle, J.A. Seed plant phylogeny and the relationships of Gnetales. Int. J. Plant Sci. 1996, 157, S3–S39. [Google Scholar] [CrossRef]
- Doyle, J.A.; Donoghue, M.J. Seed plant phylogeny and the origin of angiosperms: An experimental cladistic approach. Bot. Rev. 1986, 52, 321–431. [Google Scholar] [CrossRef]
- Kubitzki, K. Gnetaceae. In The Families and Genera of Vascular Plants; Kramer, K.U., Green, P.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 383–386. [Google Scholar]
- Hou, C.; Humphreys, A.M.; Thureborn, O.; Rydin, C. New insights into the evolutionary history of Gnetum (Gnetales). Taxon 2015, 64, 239–253. [Google Scholar] [CrossRef]
- Markgraf, F. Monographie der Gattung Gnetum Ser. 3. Bull. Jard. Bot. Buitenzorg 1930, 10, 407–511. [Google Scholar]
- Hou, C.; Wikström, N.; Strijk, J.; Rydin, C. Resolving phylogenetic relationships and species delimitations in closely related gymnosperms using high-throughput NGS, Sanger sequencing and morphology. Plant Syst. Evol. 2016, 302, 1345–1365. [Google Scholar] [CrossRef]
- Shindo, S.; Ito, M.; Ueda, K.; Kato, M.; Hasebe, M. Characterization of MADS genes in the gymnosperm Gnetum parvifolium and its implication on the evolution of reproductive organs in seed plants. Evol. Dev. 1999, 1, 180–190. [Google Scholar] [CrossRef]
- Winter, K.U.; Becker, A.; Munster, T.; Kim, J.T.; Saedler, H.; Theissen, G. MADS-box genes reveal that gnetophytes are more closely related to conifers than to flowering plants. Proc. Natl. Acad. Sci. USA 1999, 96, 7342–7347. [Google Scholar] [CrossRef]
- Becker, A.; Kaufmann, K.; Freialdenhoven, A.; Vincent, C.; Li, M.A.; Saedler, H.; Theissen, G. A novel MADS-box gene subfamily with a sister-group relationship to class B floral homeotic genes. Mol. Genet. Genomics 2002, 266, 942–950. [Google Scholar]
- Becker, A.; Saedler, H.; Theissen, G. Distinct MADS-box gene expression patterns in the reproductive cones of the gymnosperm Gnetum gnemon. Dev. Genes Evol. 2003, 213, 567–572. [Google Scholar] [CrossRef]
- Becker, A.; Winter, K.U.; Meyer, B.; Saedler, H.; Theissen, G. MADS-box gene diversity in seed plants 300 million years ago. Molec. Biol. Evol. 2000, 17, 1425–1434. [Google Scholar] [CrossRef]
- Lan, Q.; Liu, J.F.; Shi, S.Q.; Deng, N.; Jiang, Z.P.; Chang, E.M. Anatomy, microstructure and endogenous hormone changes in Gnetum parvifolium during anthesis. J. Syst. Evol. 2018, 56, 14–24. [Google Scholar] [CrossRef]
- Woodward, A.W.; Bartel, B. Auxin: Regulation, action, and interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef]
- De Smet, I.; Jürgens, G. Patterning the axis in plants–auxin in control. Curr. Opin. Genet. Dev. 2007, 17, 337–343. [Google Scholar] [CrossRef]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 373–385. [Google Scholar] [CrossRef]
- Guilfoyle, T.J. Aux/IAA proteins and auxin signal transduction. Trends Plant Sci. 1998, 3, 205–207. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.-J.; Zhang, J.-Z. Aux/IAA gene family in plants: Molecular structure, regulation, and function. Int. J. Mol. Sci. 2018, 19, 259. [Google Scholar] [CrossRef]
- Wu, W.T.; Liu, Y.X.; Wang, Y.Q.; Li, H.M.; Liu, J.X.; Tan, J.X.; He, J.D.; Bai, J.W.; Ma, H.L. Evolution analysis of the Aux/IAA gene family in plants shows dual origins and variable nuclear localization signals. Int. J. Mol. Sci. 2017, 18, 2107. [Google Scholar] [CrossRef]
- Abel, S.; Oeller, P.W.; Theologis, A. Early auxin-induced genes encode short-lived nuclear proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 326–330. [Google Scholar] [CrossRef]
- Goldfarb, B.; Lanz-Garcia, C.; Lian, Z.; Whetten, R. Aux/IAA gene family is conserved in the gymnosperm, loblolly pine (Pinus taeda). Tree Physiol. 2003, 23, 1181–1192. [Google Scholar] [CrossRef]
- Jain, M.; Kaur, N.; Garg, R.; Thakur, J.K.; Tyagi, A.K.; Khurana, J.P. Structure and expression analysis of early auxin-responsive Aux/IAA gene family in rice (Oryza sativa). Funct. Integr. Genom. 2006, 6, 47–59. [Google Scholar] [CrossRef]
- Liscum, E.; Reed, J. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 2002, 49, 387–400. [Google Scholar] [CrossRef]
- Wang, Y.J.; Deng, D.X.; Bian, Y.L.; Lv, Y.P.; Xie, Q. Genome-wide analysis of primary auxin-responsive Aux/IAA gene family in maize (Zea mays. L.). Mol. Biol. Rep. 2010, 37, 3991–4001. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715. [Google Scholar] [CrossRef]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Ann. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496. [Google Scholar] [CrossRef]
- Sherstnev, A.; Duc, C.; Cole, C.; Zacharaki, V.; Hornyik, C.; Ozsolak, F.; Milos, P.M.; Barton, G.J.; Simpson, G.G. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat. Struct. Mol. Biol. 2012, 19, 845. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Liu, M.; Downie, B.; Liang, C.; Ji, G.; Li, Q.Q.; Hunt, A.G. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2011, 108, 12533–12538. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Venu, R.; Nobuta, K.; Wu, X.; Notibala, V.; Demirci, C.; Meyers, B.C.; Wang, G.-L.; Ji, G.; Li, Q.Q. Transcriptome dynamics through alternative polyadenylation in developmental and environmental responses in plants revealed by deep sequencing. Genome Res. 2011, 21, 1478–1486. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Chua, N.H. Long noncoding RNA transcriptome of plants. J. Plant Biotechnol. 2015, 13, 319–328. [Google Scholar] [CrossRef]
- Flynn, R.A.; Chang, H.Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Ann. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.-C.; Kean, M.J.; Xu, J.; Chua, N.-H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Boerner, S.; McGinnis, K.M. Computational identification and functional predictions of long noncoding RNA in Zea mays. PLoS ONE 2012, 7, e43047. [Google Scholar] [CrossRef]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.-T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Parker, B.J.; Weiller, G.F. In Silico identification and characterization of mRNA-like noncoding transcripts in Medicago truncatula. Silico Biol. 2007, 7, 485–505. [Google Scholar]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.-Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.X.; Mei, W.B.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT-and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [Green Version]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef] [PubMed]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, 297–299. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, 369–373. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Albert, V.A.; Barbazuk, W.B.; Der, J.P.; Leebens-Mack, J.; Ma, H.; Palmer, J.D.; Rounsley, S.; Sankoff, D.; Schuster, S.C.; Soltis, D.E. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.F.; Banf, M.; Dai, X.B.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Li, J.W.; Ma, W.; Zeng, P.; Wang, J.Y.; Geng, B.; Yang, J.C.; Cui, Q.H. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Froberg, J.E.; Lee, J.T. Long noncoding RNAs: Fresh perspectives into the RNA world. Trends Biochem. Sci. 2014, 39, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Liu, Z.-M.; Liao, Y.-Y.; Yang, M.; Leitch, A.R.; Leitch, I.J.; Van de Peer, Y.; Leebens-Mack, J.H.; Song, C.; Hou, C.; et al. The Welwitschia genome reveals new insights into the early evolutionary history of seed plants. Unpublished.
- Guan, R.; Zhao, Y.P.; Zhang, H.; Fan, G.Y.; Liu, X.; Zhou, W.B.; Shi, C.C.; Wang, J.H.; Liu, W.Q.; Liang, X.M.; et al. Draft genome of the living fossil Ginkgo biloba. Gigascience 2016, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jones, B.; Li, Z.; Frasse, P.; Delalande, C.; Regad, F.; Chaabouni, S.; Latche, A.; Pech, J.-C.; Bouzayen, M. The tomato Aux/IAA transcription factor IAA9 is involved in fruit development and leaf morphogenesis. Plant Cell 2005, 17, 2676–2692. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing Version 3.2.0; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 2 October 2018).
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Hou, C.; Saunders, R.M.; Deng, N.; Wan, T.; Su, Y. Pollination drop proteome and reproductive organ transcriptome comparison in Gnetum reveals entomophilous adaptation. Genes 2019, 10, 800. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Sang, Y.; Liu, X.; Xing, S.; Li, J.; Tang, H.; Sun, L. Transcriptome profile analysis from different sex types of Ginkgo biloba L. Front. Plant. Sci. 2016, 7, 871. [Google Scholar] [CrossRef] [Green Version]
- Pirone-Davies, C.; Prior, N.; von Aderkas, P.; Smith, D.; Hardie, D.; Friedman, W.E.; Mathews, S. Insights from the pollination drop proteome and the ovule transcriptome of Cephalotaxus at the time of pollination drop production. Ann. Bot. 2016, 117, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.-B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [Green Version]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.-K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, G.; Reddy, A. Regulation of alternative splicing of pre-mRNAs by stresses. In Nuclear pre-mRNA Processing in Plants; Reddy, A.S., Golovkin, M., Eds.; Springer: Heidelberg, Germany, 2008; pp. 257–275. [Google Scholar]
- Tai, H.H.; Williams, M.; Iyengar, A.; Yeates, J.; Beardmore, T. Regulation of the β-hydroxyacyl ACP dehydratase gene of Picea mariana by alternative splicing. Plant Cell Rep. 2007, 26, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Cheng, H.; Cai, R.; Li, L.L.; Chang, J.; Zhu, J.; Zhang, F.X.; Chen, L.J.; Wang, Y.; Cheng, S.H. Molecular cloning and function analysis of an anthocyanidin synthase gene from Ginkgo biloba, and its expression in abiotic stress responses. Mol. Cells 2008, 26, 536–547. [Google Scholar] [PubMed]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18. [Google Scholar] [CrossRef]
- Xing, D.; Li, Q.Q. Alternative polyadenylation and gene expression regulation in plants. Wiley Interdiscip. Rev. RNA 2011, 2, 445–458. [Google Scholar] [CrossRef]
- Shen, Y.; Ji, G.; Haas, B.J.; Wu, X.; Zheng, J.; Reese, G.J.; Li, Q.Q. Genome level analysis of rice mRNA 3′-end processing signals and alternative polyadenylation. Nucleic Acids Res. 2008, 36, 3150–3161. [Google Scholar] [CrossRef] [Green Version]
- Chinn, E.; Silverthorne, J.; Hohtola, A. Light-regulated and organ-specific expression of types 1, 2, and 3 light-harvesting complex b mRNAs in Ginkgo biloba. Plant Physiol. 1995, 107, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Simpson, G.G.; Dijkwel, P.P.; Quesada, V.; Henderson, I.; Dean, C. FY is an RNA 3′ end-processing factor that interacts with FCA to control the Arabidopsis floral transition. Cell 2003, 113, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Marquardt, S.; Lister, C.; Swiezewski, S.; Dean, C. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 2010, 327, 94–97. [Google Scholar] [CrossRef]
- Ye, J.; Cheng, S.; Zhou, X.; Chen, Z.; Kim, S.U.; Tan, J.; Zheng, J.; Xu, F.; Zhang, W.; Liao, Y. A global survey of full-length transcriptome of Ginkgo biloba reveals transcript variants involved in flavonoid biosynthesis. Ind. Crops Prod. 2019, 139, 111547. [Google Scholar] [CrossRef]
- Jansson, S.; Gustafsson, P. Type I and type II genes for the chlorophyll a/b-binding protein in the gymnosperm Pinus sylvestris (Scots pine): cDNA cloning and sequence analysis. Plant Mol. Biol. 1990, 14, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.Z.; Shen, G.A.; Liu, C.H.; Liu, X.J.; Tan, F.; Sun, X.F.; Tang, K.X. Molecular cloning and sequence analysis of a novel chalcone synthase cDNA from Ginkgo biloba. DNA Seq. 2004, 15, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Xia, X.; Jiang, H.; Lu, Z.; Cui, J.; Cao, F.; Jin, B. Genome-wide identification and characterization of novel lncRNAs in Ginkgo biloba. Trees 2018, 32, 1429–1442. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Chanderbali, A.S.; Yoo, M.-J.; Zahn, L.M.; Brockington, S.F.; Wall, P.K.; Gitzendanner, M.A.; Albert, V.A.; Leebens-Mack, J.; Altman, N.S.; Ma, H. Conservation and canalization of gene expression during angiosperm diversification accompany the origin and evolution of the flower. Proc. Natl. Acad. Sci. USA 2010, 107, 22570–22575. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Melzer, R.; Theissen, G. Molecular interactions of orthologues of floral homeotic proteins from the gymnosperm Gnetum gnemon provide a clue to the evolutionary origin of ‘floral quartets’. Plant J. 2010, 64, 177–190. [Google Scholar] [CrossRef]
- Hou, C.; Li, L.; Liu, Z.; Su, Y.; Wan, T. Diversity and expression patterns of MADS-box genes in Gnetum luofuense—Implication in functional diversity and evolution. Unpublished.
- Farrona, S.; Hurtado, L.; Bowman, J.L.; Reyes, J.C. The Arabidopsis thaliana SNF2 homolog AtBRM controls shoot development and flowering. Development 2004, 131, 4965–4975. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.X.; Deng, L.L.; Zeng, K.F. Genome-wide in silico identification of membrane-bound transcription factors in plant species. Peerj 2017, 5, e4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.I.; Muthusamy, M.; Nawaz, M.A.; Hong, J.K.; Lim, M.-H.; Kim, J.A.; Jeong, M.-J. Genome-wide analysis of spatiotemporal gene expression patterns during floral organ development in Brassica rapa. Mol. Genet. Genom. 2019, 294, 1403–1420. [Google Scholar] [CrossRef] [PubMed]
- Szecsi, J.; Joly, C.; Bordji, K.; Varaud, E.; Cock, J.M.; Dumas, C.; Bendahmane, M. BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J. 2006, 25, 3912–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heisler, M.G.; Atkinson, A.; Bylstra, Y.H.; Walsh, R.; Smyth, D.R. SPATULA, a gene that controls development of carpel margin tissues in Arabidopsis, encodes a bHLH protein. Development 2001, 128, 1089–1098. [Google Scholar]
- Gish, L.A.; Clark, S.E. The RLK/Pelle family of kinases. Plant J. 2011, 66, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.E.; Running, M.P.; Meyerowitz, E.M. CLAVATA1, a regulator of meristem and flower development in Arabidopsis. Development 1993, 119, 397–418. [Google Scholar]
- Clark, S.E.; Williams, R.W.; Meyerowitz, E.M. The CLAVATA1 gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis. Cell 1997, 89, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Avila, C.; Pérez-Rodríguez, J.; Canovas, F. Molecular characterization of a receptor-like protein kinase gene from pine (Pinus sylvestris L.). Planta 2006, 224, 12–19. [Google Scholar] [CrossRef]
- Ljung, K. Auxin metabolism and homeostasis during plant development. Development 2013, 140, 943–950. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Different developmental stages of female strobili in Gnetum luofuense C.Y. Cheng.

Figure 2.
(A) Numbers and proportion of varied isoforms generated by PacBio single-molecular long-read technology (SMRT) sequencing; (B) Length distribution and accumulation proportion of consensus isoforms; (C) Length distribution of assembled full-length transcripts; (D) Percentages of varied types of Benchmarking Universal Single Copy Orthologs (BUSCO) orthologues, where F01 represents the full-length transcriptome generated in the present study.
Figure 2.
(A) Numbers and proportion of varied isoforms generated by PacBio single-molecular long-read technology (SMRT) sequencing; (B) Length distribution and accumulation proportion of consensus isoforms; (C) Length distribution of assembled full-length transcripts; (D) Percentages of varied types of Benchmarking Universal Single Copy Orthologs (BUSCO) orthologues, where F01 represents the full-length transcriptome generated in the present study.

Figure 3.
(A) All gene and novel gene annotation by the searching of seven databases; (B) NR (NCBI nonredundant protein sequences) homologous species distribution diagram of all genes and novel genes; (C) Distribution of GO (Gene Ontology) terms for all annotated genes and novel genes, cellular component and molecular function; (D) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways of all genes and novel genes involved in cellular process, environmental information processing, genetic information processing, organism systems, and metabolism.
Figure 3.
(A) All gene and novel gene annotation by the searching of seven databases; (B) NR (NCBI nonredundant protein sequences) homologous species distribution diagram of all genes and novel genes; (C) Distribution of GO (Gene Ontology) terms for all annotated genes and novel genes, cellular component and molecular function; (D) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways of all genes and novel genes involved in cellular process, environmental information processing, genetic information processing, organism systems, and metabolism.

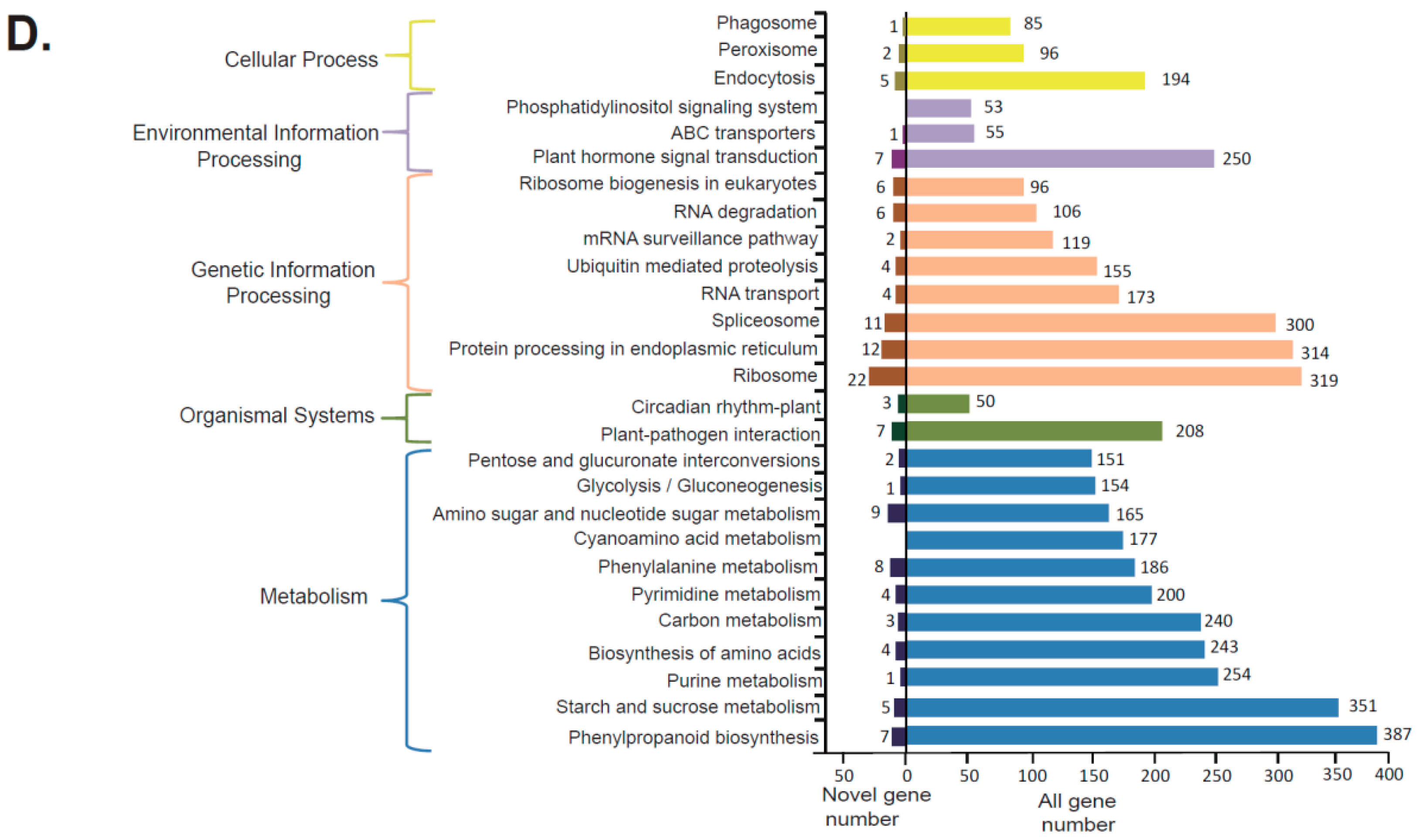
Figure 4.
(A) A schematic graph and distribution of varied alternative splicing (AS) events found in the female strobili of G. luofuense; (B) Distribution of the number of poly (A) sites (x-axis) associated with gene numbers (y-axis); (C) The nucleotide composition around poly (A) cleavage sites was presented with the relative frequency of a nucleotide (y-axis) and transcriptome composition cross all poly (A) cleavage sites (x-axis); (D) three over-represented motifs at 25 nts upstream of the poly (A) site for all G. luofuense full-length transcripts.
Figure 4.
(A) A schematic graph and distribution of varied alternative splicing (AS) events found in the female strobili of G. luofuense; (B) Distribution of the number of poly (A) sites (x-axis) associated with gene numbers (y-axis); (C) The nucleotide composition around poly (A) cleavage sites was presented with the relative frequency of a nucleotide (y-axis) and transcriptome composition cross all poly (A) cleavage sites (x-axis); (D) three over-represented motifs at 25 nts upstream of the poly (A) site for all G. luofuense full-length transcripts.
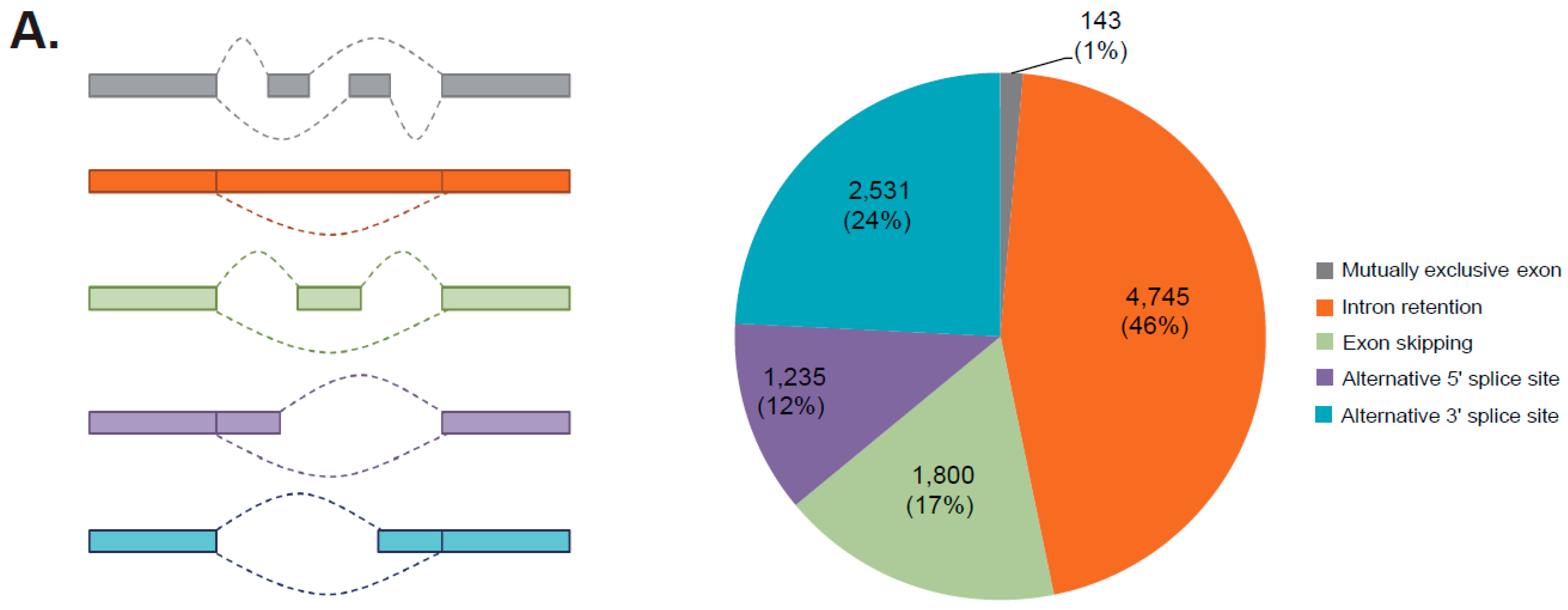
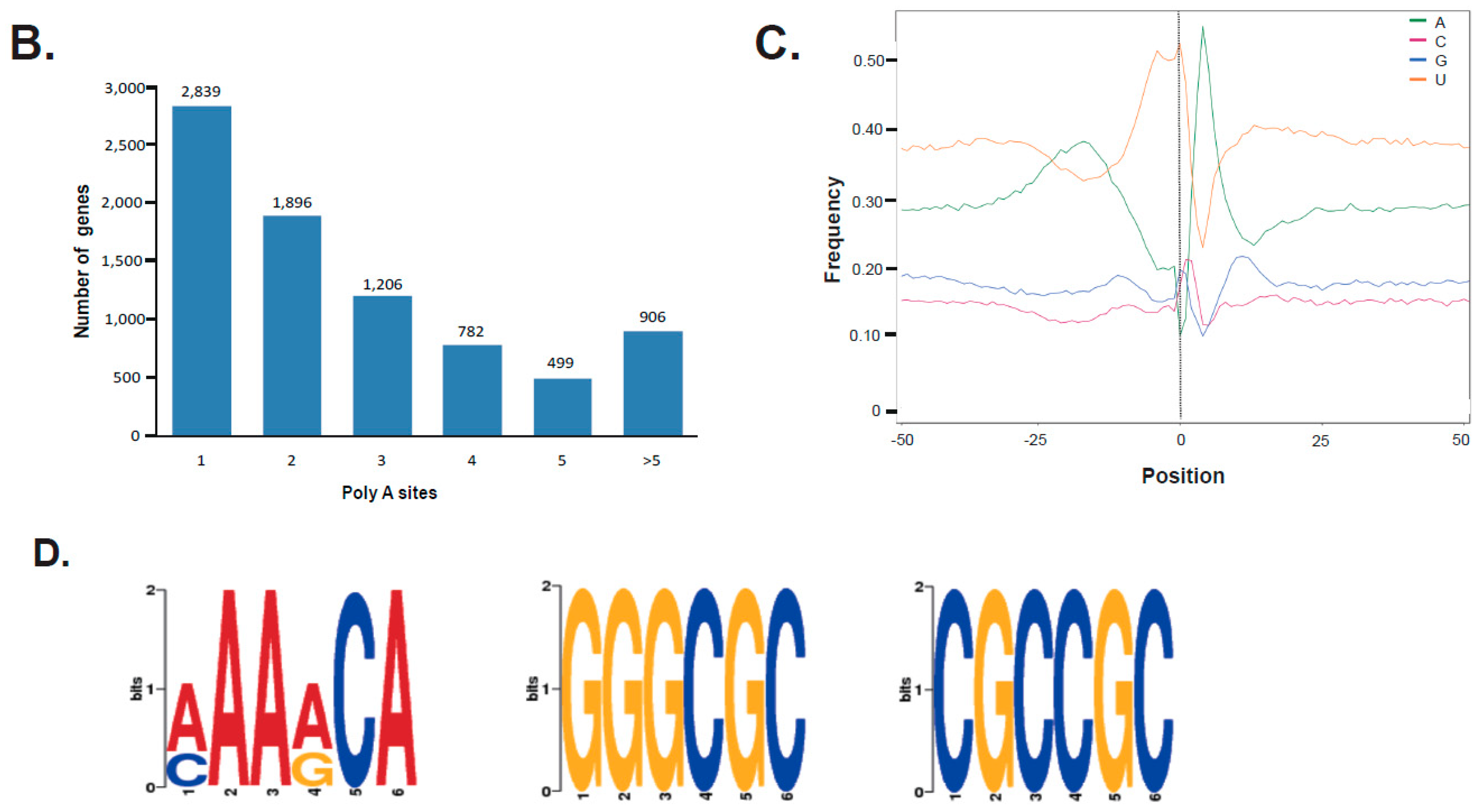
Figure 5.
(A) Length distribution of open reading frames (ORFs) detected based on assembled full-length transcripts; (B) Top 30 numbers of protein regulators including transcript regulators (TRs), transcription factors (TFs), and protein kinases (PKs); (C) A Venn diagram exhibiting the number of lncRNAs identified by four different approaches: CPC (Coding Potential Calculator), CNCI (Coding-Non-Coding Index), CPAT (Coding Potential Assessment Tool), and Pfam (Protein Family); (D) Functional classification and numbers of major types of lncRNAs.
Figure 5.
(A) Length distribution of open reading frames (ORFs) detected based on assembled full-length transcripts; (B) Top 30 numbers of protein regulators including transcript regulators (TRs), transcription factors (TFs), and protein kinases (PKs); (C) A Venn diagram exhibiting the number of lncRNAs identified by four different approaches: CPC (Coding Potential Calculator), CNCI (Coding-Non-Coding Index), CPAT (Coding Potential Assessment Tool), and Pfam (Protein Family); (D) Functional classification and numbers of major types of lncRNAs.
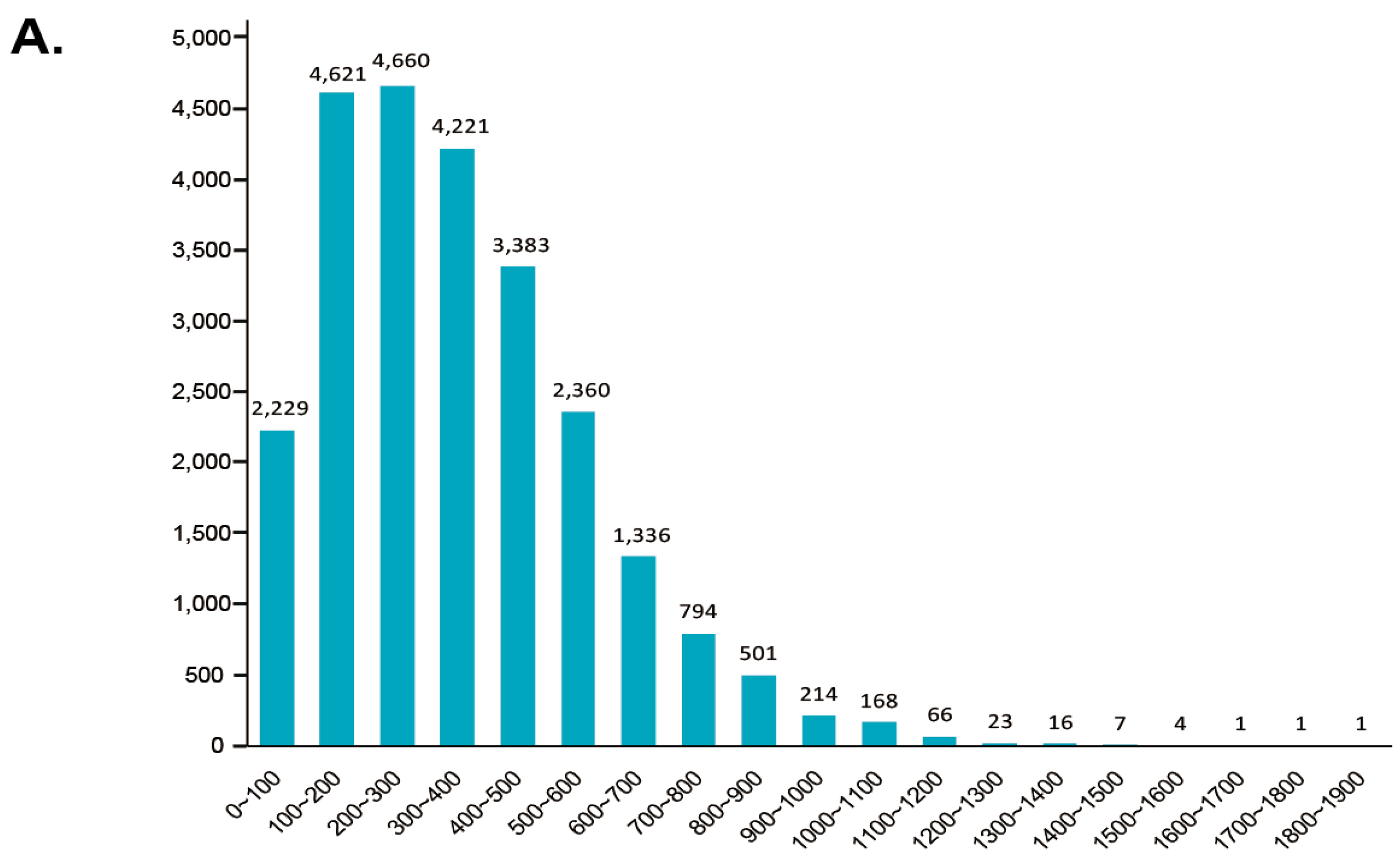

Figure 6.
(A) The expression of six Aux/IAA genes from G. luofuense verified by qRT-PCR; the relative expression values normalized with the ΔΔCt-method are presented, above and qRT-PCR products were exhibited by gel electrophoresis, as shown below; (B) Molecular evolutionary rates of Aux/IAAs represented by paralogous gene pairs of Ka/Ks values in 11 land plant species; (C) Phylogenetic relationship among canonical Aux/IAA genes for seed plants (left), and varied types of structures examined (right) and the content of conservative amino acids, corresponding to five motifs and four conservative domains, i.e., Domains I–IV, was expressed by the height of characters (below).
Figure 6.
(A) The expression of six Aux/IAA genes from G. luofuense verified by qRT-PCR; the relative expression values normalized with the ΔΔCt-method are presented, above and qRT-PCR products were exhibited by gel electrophoresis, as shown below; (B) Molecular evolutionary rates of Aux/IAAs represented by paralogous gene pairs of Ka/Ks values in 11 land plant species; (C) Phylogenetic relationship among canonical Aux/IAA genes for seed plants (left), and varied types of structures examined (right) and the content of conservative amino acids, corresponding to five motifs and four conservative domains, i.e., Domains I–IV, was expressed by the height of characters (below).
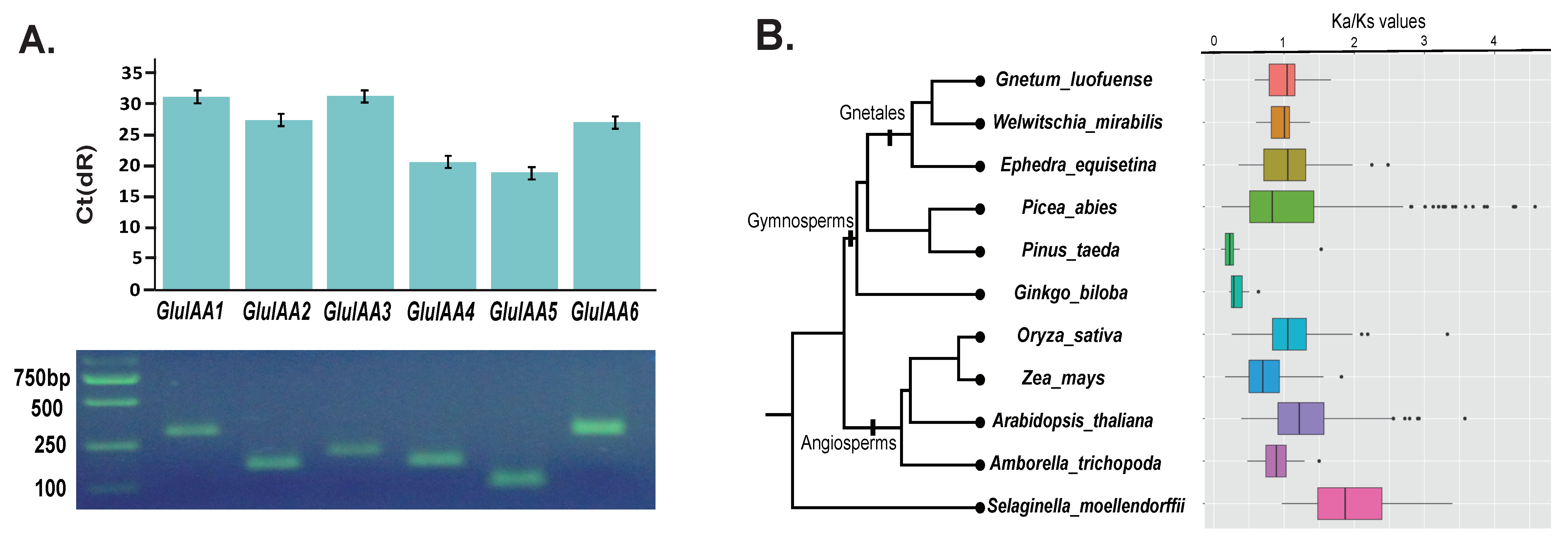

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hou, C.; Deng, N.; Su, Y. PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales). Forests 2019, 10, 1043. https://doi.org/10.3390/f10111043
AMA Style
Hou C, Deng N, Su Y. PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales). Forests. 2019; 10(11):1043. https://doi.org/10.3390/f10111043
Chicago/Turabian StyleHou, Chen, Nan Deng, and Yingjuan Su. 2019. "PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales)" Forests 10, no. 11: 1043. https://doi.org/10.3390/f10111043
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.