Assessment of Genetic Diversity and Population Genetic Structure of Norway Spruce (Picea abies (L.) Karsten) at Its Southern Lineage in Europe. Implications for Conservation of Forest Genetic Resources

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
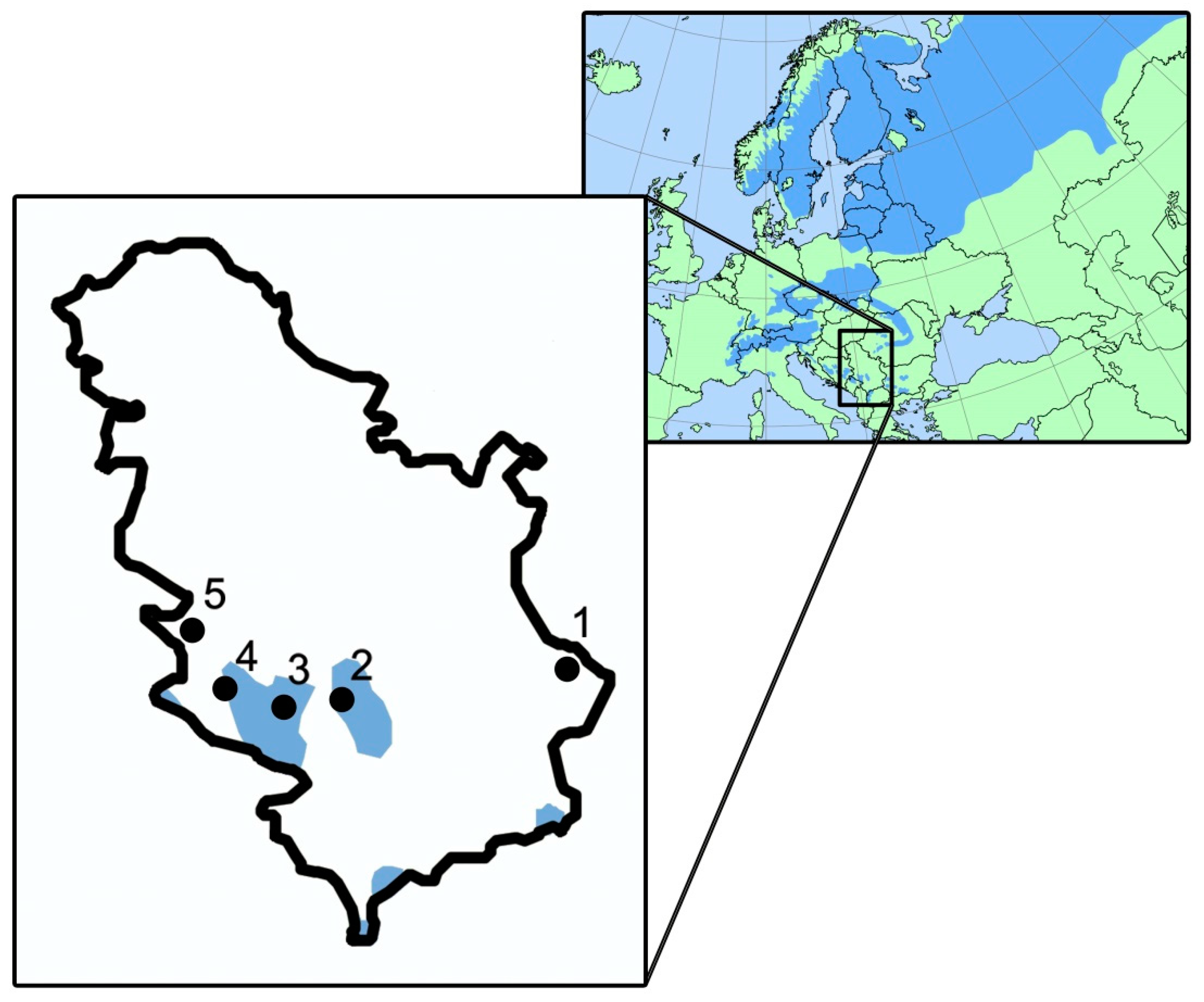
2.1. Plant Material and DNA Isolation
2.2. EST-SSR Data Analysis
2.3. Data Analysis
2.3.1. EST-SSR Variation and Genetic Diversity within Populations
2.3.2. Genetic Divergence between Populations
3. Results
3.1. EST-SSR Variation and Genetic Diversity within Populations
3.2. Genetic Variation within and between Populations
4. Discussion
Conservation of Norway Spruce Forest Genetic Resources in Serbia
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tollefsrud, M.M.; Kissling, R.; Gugerli, F.; Johnsen, Ø.; Skrøppa, T.; Cheddadi, R.; van der Knaap, W.O.; Latałowa, M.; TerHürne-Berson, R.; Litt, T. Genetic consequences of glacial survival and postglacial colonization in norway spruce: Combined analysis of mitochondrial DNA and fossil pollen. Mol. Ecol. 2008, 17, 4134–4150. [Google Scholar] [CrossRef]
- Tsuda, Y.; Chen, J.; Stocks, M.; Källman, T.; Sønstebø, J.H.; Parducci, L.; Semerikov, V.; Sperisen, C.; Politov, D.; Ronkainen, T. The extent and meaning of hybridization and introgression between siberian spruce (Picea obovata) and norway spruce (Picea abies): Cryptic refugia as stepping stones to the west? Mol. Ecol. 2016, 25, 2773–2789. [Google Scholar] [CrossRef] [PubMed]
- Sperisen, C.; Büchler, U.; Mátyás, G. Genetic variation of mitochondrial DNA reveals subdivision of norway spruce (Picea abies (L.) Karst.). In Molecular Tools for Screening Biodiversity; Springer: Berlin, Germany, 1998; pp. 413–417. [Google Scholar]
- Ravazzi, C. Late quaternary history of spruce in Southern Europe. Rev. Palaeobot. Palynol. 2002, 120, 131–177. [Google Scholar] [CrossRef]
- Latałowa, M.; van der Knaap, W.O. Late quaternary expansion of norway spruce Picea abies (L.) Karst. in Europe according to pollen data. Quat. Sci. Rev. 2006, 25, 2780–2805. [Google Scholar] [CrossRef]
- Kesić, L.; Matović, B.; Stojnić, S.; Stjepanović, S.; Stojanović, D. Climate change as a factor reducing the growth of trees in the pure norway spruce stand (Picea abies (L.) H. Karst.) in the National park “Kopaonik”. Topola/Poplar 2016, 197–198, 25–34. [Google Scholar]
- Marković, Č.; Stojanović, A. Differences in bark beetle (Ips typographus and Pityogenes chalcographus) abundance in a strict spruce reserve and the surrounding spruce forests of Serbia. Phytoparasitica 2010, 38, 31–37. [Google Scholar] [CrossRef]
- Lindner, M.; Maroschek, M.; Netherer, S.; Kremer, A.; Barbati, A.; Garcia-Gonzalo, J.; Seidl, R.; Delzon, S.; Corona, P.; Kolström, M. Climate change impacts, adaptive capacity, and vulnerability of European forest ecosystems. For. Ecol. Manag. 2010, 259, 698–709. [Google Scholar] [CrossRef]
- Keren, S.; Motta, R.; Govedar, Z.; Lucic, R.; Medarevic, M.; Diaci, J. Comparative structural dynamics of the janj mixed old-growth mountain forest in Bosnia and Herzegovina: Are conifers in a long-term decline? Forests 2014, 5, 1243–1266. [Google Scholar] [CrossRef]
- Avtzis, D.; Aravanopoulos, F. Host tree and insect genetic diversity on the borderline of natural distribution: A case study of Picea abies and Pityogenes chalcographus (Coleoptera, Scolytinae) in Greece. Silva Fenn. 2011, 45, 157–164. [Google Scholar] [CrossRef]
- Stojanović, D.B.; Matović, B.; Orlović, S.; Kržič, A.; Trudić, B.; Galić, Z.; Stojnić, S.; Pekeč, S. Future of the main important forest tree species in Serbia from the climate change perspective. SEEFOR 2014, 5, 117–124. [Google Scholar] [CrossRef]
- Belletti, P.; Ferrazzini, D.; Ducci, F.; De Rogatis, A.; Mucciarelli, M. Genetic diversity of Italian populations of Abies alba. Dendrobiology 2017, 77, 147–159. [Google Scholar] [CrossRef]
- Markert, J.A.; Champlin, D.M.; Gutjahr-Gobell, R.; Grear, J.S.; Kuhn, A.; McGreevy, T.J.; Roth, A.; Bagley, M.J.; Nacci, D.E. Population genetic diversity and fitness in multiple environments. BMC Evol. Biol. 2010, 10, 205. [Google Scholar] [CrossRef]
- Koskela, E.; Ollikainen, M.; Pukkala, T. Biodiversity conservation in commercial boreal forestry: The optimal rotation age and retention tree volume. For. Sci. 2007, 53, 443–452. [Google Scholar]
- Fady, B.; Aravanopoulos, F.A.; Alizoti, P.; Mátyás, C.; von Wühlisch, G.; Westergren, M.; Belletti, P.; Cvjetkovic, B.; Ducci, F.; Huber, G.; et al. Evolution-based approach needed for the conservation and silviculture of peripheral forest tree populations. For. Ecol. Manag. 2016, 375, 66–75. [Google Scholar] [CrossRef]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef]
- Gupta, P.; Rustgi, S.; Sharma, S.; Singh, R.; Kumar, N.; Balyan, H. Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol. Genet. Genomics 2003, 270, 315–323. [Google Scholar] [CrossRef]
- Hu, J.; Wang, L.; Li, J. Comparison of genomic SSR and EST-SSR markers for estimating genetic diversity in cucumber. Biol. Plant. 2011, 55, 577–580. [Google Scholar] [CrossRef]
- Feng, S.; Li, W.; Huang, H.; Wang, J.; Wu, Y. Development, characterization and cross-species/genera transferability of EST-SSR markers for rubber tree (Hevea brasiliensis). Mol. Breed. 2009, 23, 85–97. [Google Scholar] [CrossRef]
- Fluch, S.; Burg, A.; Kopecky, D.; Homolka, A.; Spiess, N.; Vendramin, G.G. Characterization of variable EST SSR markers for norway spruce (Picea abies L.). BMC Res. Notes 2011, 4, 401. [Google Scholar] [CrossRef]
- Dumolin, S.; Demesure, B.; Petit, R. Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient pcr method. Theor. Appl. Genet. 1995, 91, 1253–1256. [Google Scholar] [CrossRef]
- Skrøppa, T. EUFORGEN Technical Guidelines for Genetic Conservation and Use for Norway Spruce (Picea abies); Bioversity International: Rome, Italy, 2003. [Google Scholar]
- Yeh Francis, C.; Yang, R.; Boyle Timothy, B.; Ye, Z.; Mao Judy, X. POPGENE Version 1.32, the User-Friendly Shareware for Population Genetic Analysis; Molecular Biology and Biotechnology Centre, University of Alberta: Alberta, AB, Canada, 1999; Available online: http://www.ualbertaca/~fyeh/ (accessed on 6 December 2018).
- Kalinowski, S.T. HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Resour. 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Dakin, E.; Avise, J. Microsatellite null alleles in parentage analysis. Heredity 2004, 93, 504. [Google Scholar] [CrossRef]
- Chapuis, M.-P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2006, 24, 621–631. [Google Scholar] [CrossRef]
- Raymond, M. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Michalakis, Y.; Excoffier, L. A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 1996, 142, 1061–1064. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Resour. 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar]
- Takezaki, N.; Nei, M.; Tamura, K. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol. Biol. Evol. 2009, 27, 747–752. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Gutkowska, J.; Borys, M.; Tereba, A.; Tkaczyk, M.; Oszako, T.; Nowakowska, J.A. Genetic variability and health of norway spruce stands in the Regional Directorate of the State Forests in Krosno. For. Res. Pap. 2017, 78, 56–66. [Google Scholar] [CrossRef]
- Meloni, M.; Perini, D.; Binelli, G. The distribution of genetic variation in norway spruce (Picea abies Karst.) populations in the Western Alps. J. Biogeogr. 2007, 34, 929–938. [Google Scholar] [CrossRef]
- Radu, R.G.; Curtu, L.A.; Spârchez, G. Genetic diversity of norway spruce [Picea abies (L.) Karst.] in romanian carpathians. Ann. For. Res. 2014, 57, 19. [Google Scholar] [CrossRef]
- Westergren, M.; Bozic, G.; Kraigher, H. Genetic diversity of core vs. peripheral norway spruce native populations at a local scale in Slovenia. iForest 2018, 11, 104. [Google Scholar] [CrossRef]
- Máchová, P.; Trčková, O.; Cvrčková, H. Use of nuclear microsatellite loci for evaluating genetic diversity of selected populations of Picea abies (L.) Karsten in the Czech Republic. Forests 2018, 9, 92. [Google Scholar] [CrossRef]
- Unger, G.; Konrad, H.; Geburek, T. Does spatial genetic structure increase with altitude? An answer from Picea abies in Tyrol, Austria. Plant Syst. Evol. 2011, 292, 133–141. [Google Scholar] [CrossRef]
- Cvjetković, B.; Konnert, M.; Fussi, B.; Mataruga, M.; Šijačić-Nikolić, M.; Daničić, V.; Lučić, A. Norway spruce (Picea abies Karst.) variability in progeny tests in Bosnia and Herzegovina. Genetika 2017, 49, 259–272. [Google Scholar] [CrossRef]
- Scotti, I.; Paglia, G.; Magni, F.; Morgante, M. Population genetics of norway spruce (Picea abies Karst.) at regional scale: Sensitivity of different microsatellite motif classes in detecting differentiation. Ann. For. Sci. 2006, 63, 485–491. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. In Population Genetics of Forest Trees; Springer: Berlin, Germany, 1992; pp. 95–124. [Google Scholar]
- Porth, I.; El-Kassaby, Y.A. Assessment of the genetic diversity in forest tree populations using molecular markers. Diversity 2014, 6, 283–295. [Google Scholar] [CrossRef]
- Nowakowska, J.A.; Zachara, T.; Konecka, A. Genetic variability of scots pine (Pinus sylvestris L.) and norway spruce (Picea abies L. Karst.) natural regeneration compared with their maternal stands. For. Res. Pap. 2014, 75, 47–54. [Google Scholar] [CrossRef]
- Petit, R.J.; El Mousadik, A.; Pons, O. Identifying populations for conservation on the basis of genetic markers. Conserv. Biol. 1998, 12, 844–855. [Google Scholar] [CrossRef]
- Pandey, M.; Rajora, O.P. Higher fine-scale genetic structure in peripheral than in core populations of a long-lived and mixed-mating conifer-eastern white cedar (Thuja occidentalis L.). BMC Evol. Biol. 2012, 12, 48. [Google Scholar] [CrossRef]
- Ganopoulos, I.; Aravanopoulos, F.A.; Argiriou, A.; Kalivas, A.; Tsaftaris, A. Is the genetic diversity of small scattered forest tree populations at the southern limits of their range more prone to stochastic events? A wild cherry case study by microsatellite-based markers. Tree Genet. Genomes 2011, 7, 1299–1313. [Google Scholar] [CrossRef]
- Checker, V.G.; Khurana, P. Molecular and functional characterization of mulberry EST encoding remorin (MiREM) involved in abiotic stress. Plant Cell Rep. 2013, 32, 1729–1741. [Google Scholar] [CrossRef]
- La Camera, S.; L’Haridon, F.; Astier, J.; Zander, M.; Abou-Mansour, E.; Page, G.; Thurow, C.; Wendehenne, D.; Gatz, C.; Métraux, J.P. The glutaredoxin ATGRXS13 is required to facilitate Botrytis cinerea infection of Arabidopsis thaliana plants. Plant J. 2011, 68, 507–519. [Google Scholar] [CrossRef]
- Piotti, A.; Leonardi, S.; Piovani, P.; Scalfi, M.; Menozzi, P. Spruce colonization at treeline: Where do those seeds come from? Heredity 2009, 103, 136. [Google Scholar] [CrossRef]
- Slatkin, M. A measure of population subdivision based on microsatellite allele frequencies. Genetics 1995, 139, 457–462. [Google Scholar]
- Shi, X.; Wen, Q.; Cao, M.; Guo, X.; Xu, L.-A. Genetic diversity and structure of natural quercus variabilis population in china as revealed by microsatellites markers. Forests 2017, 8, 495. [Google Scholar] [CrossRef]
- Tollefsrud, M.M.; Sønstebø, J.H.; Brochmann, C.; Johnsen, Ø.; Skrøppa, T.; Vendramin, G.G. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European Picea abies. Heredity 2009, 102, 549. [Google Scholar] [CrossRef]
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Aravanopoulos, F. Genetic monitoring in natural perennial plant populations. Botany 2011, 89, 75–81. [Google Scholar] [CrossRef]
- Bashalkhanov, S.; Pandey, M.; Rajora, O.P. A simple method for estimating genetic diversity in large populations from finite sample sizes. BMC Genet. 2009, 10, 84. [Google Scholar] [CrossRef]
- Jacquemyn, H.; Roldán-Ruiz, I.; Honnay, O. Evidence for demographic bottlenecks and limited gene flow leading to low genetic diversity in a rare thistle. Conserv. Genet. 2010, 11, 1979–1987. [Google Scholar] [CrossRef]
- Murray, B.; Young, A. Widespread chromosome variation in the endangered grassland forb Rutidosis leptorrhynchoides F. Muell. (Asteraceae: Gnaphalieae). Ann. Bot.-Lond. 2001, 87, 83–90. [Google Scholar] [CrossRef]
- Tomimatsu, H.; Ohara, M. Genetic diversity and local population structure of fragmented populations of Trillium camschatcense (Trilliaceae). Biol. Conserv. 2003, 109, 249–258. [Google Scholar] [CrossRef]
- De Vries, S.; Alan, M.; Bozzano, M.; Burianek, V.; Collin, E.; Cottrell, J.; Ivankovic, M.; Kelleher, C.; Koskela, J.; Rotach, P. Pan-European strategy for genetic conservation of forest trees and establishment of a core network of dynamic conservation units. In European Forest Genetic Resources Programme (EUFORGEN); Bioversity International: Rome, Italy, 2015. [Google Scholar]
- Matović, B.S.D.; Kesić, L.; Stjepanović, S. Uticaj klime na prirast i vitalnost smrče na Kopaoniku. Topola/Poplar 2018, 201/202, 99–116. [Google Scholar]
- Griess, V.C.; Acevedo, R.; Härtl, F.; Staupendahl, K.; Knoke, T. Does mixing tree species enhance stand resistance against natural hazards? A case study for spruce. For. Ecol. Manag. 2012, 267, 284–296. [Google Scholar] [CrossRef]
- Zong, J.-W.; Zhao, T.-T.; Ma, Q.-H.; Liang, L.-S.; Wang, G.-X. Assessment of genetic diversity and population genetic structure of Corylus mandshurica in China using SSR markers. PLoS ONE 2015, 10, e0137528. [Google Scholar] [CrossRef]
- Koski, V.; Skrøppa, T.; Paule, L.; Wolf, H.; Turok, J. Technical Guidelines for Genetic Conservation of Norway Spruce Picea abies (L.) Karst.); Bioversity International: Rome, Italy, 1997. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| No | Population | Mountain Range | Latitude (dd.mm) | Longitude (dd.mm) | Altitude (m a.s.l.) | Mean Air Temperature (°C ) | Annual Sum of Precipitation (mm) |
|---|---|---|---|---|---|---|---|
| 1 | Stara Planina | Balkan Mt | 43°18′ | 22°47′ | 1300 | 7.5 | 634 |
| 2 | Kopaonik | Dinaric Alps | 43°18′ | 20°46′ | 1530 | 6.5 | 753 |
| 3 | Golija | Dinaric Alps | 43°18′ | 20°17′ | 1650 | 6.0 | 757 |
| 4 | Zlatar | Dinaric Alps | 43°26′ | 19°49′ | 1200 | 6.4 | 739 |
| 5 | Tara | Dinaric Alps | 43°53′ | 19°31′ | 1000 | 8.6 | 823 |
| Locus | Primer Sequences (5′-3′) | Repeat Motif | Fragment Length (bp) | Ta (°C) |
|---|---|---|---|---|
| WS00716.F13 1 | TCAAGTAATGGACAAACGATACA TTTCCAATAGAATGGTGGATTT | (GA)10 | 214–320 | 53 |
| WS0092.M15 1 | GATGTTGCAGGCATTCAGAG GCACCAGCTCGATTGACTA | (TCC)6 | 204–216 | 53 |
| WS0022.B15 1 | TTTGTAGGTGCTGCAGAGAT TGGCTTTTATTCCAGCAAGA | (AG)12 | 160–220 | 53 |
| WS0073.H08 1 | TGCTCTCTTATTCGGGCTTC AAGAACAAGGTTCCCAATG | (AT)14 | 209–265 | 55 |
| WS00111.K13 1 | GACTGAAGATGCCGATATGC GGCCATATCTCAAAATAAAGAA | (AT)9 | 181–237 | 55 |
| WS0023.B03 1 | AGCAFGTGGGGTCAAAGTT AAAGAAAGCATGCATATGACTCAG | (AT)10 | 170–224 | 62 |
| Pa44 2 | AAGGCAGCAAAGTGAGAA CTTGGCATTCCCTAGTGAGC | (GGA)n | 271–305 | 62 |
| Pa51 2 | CAGATGTGGGCACTTGTTTG TGGTCATGGTGGTGTTCAT | (CCA)n | 124–145 | 62 |
| Locus | Na | Ne | I | Ho | He | Fst | Fis | fn |
|---|---|---|---|---|---|---|---|---|
| WS00716.F13 | 22 | 15.280 | 2.836 | 0.060 | 0.934 | 0.016 | −0.015 | 0.003 |
| WS0092.M15 | 3 | 1.677 | 0.666 | 0.473 | 0.403 | 0.000 | −0.301 | 0.000 |
| WS0022.B15 | 17 | 9.179 | 2.431 | 0.194 | 0.891 | 0.011 | 0.090 | 0.218 |
| WS0073.H08 | 6 | 3.442 | 1.409 | 0.317 | 0.709 | 0.000 | 0.045 | 0.003 |
| WS00111.K13 | 29 | 22.114 | 3.191 | 0.081 | 0.954 | 0.012 | 0.031 | 0.021 |
| WS0023.B03 | 26 | 19.299 | 3.074 | 0.397 | 0.948 | 0.002 | 0.365 * | 0.177 |
| Pa44 | 8 | 1.194 | 0.417 | 0.827 | 0.162 | 0.000 | −0.007 | 0.000 |
| Pa51 | 2 | 1.034 | 0.085 | 0.966 | 0.033 | 0.000 | −0.050 | 0.782 |
| Mean | 14.12 | 9.152 | 1.764 | 0.414 | 0.629 | 0.007 | 0.067 | 0.154 |
| St. Dev. | 8.67 | 1.271 | 0.332 | 0.377 |
| Population | Ne | He | I | AR | pAR | Fis | Fis after Null Allele Correction | Fst |
|---|---|---|---|---|---|---|---|---|
| Stara Planina | 6.621 | 0.612 | 1.574 | 9.86 | 0.52 | 0.091 | −0.441 | |
| Kopaonik | 6.462 | 0.617 | 1.587 | 9.70 | 0.13 | 0.074 | 0.002 | |
| Golija | 7.984 | 0.635 | 1.706 | 11.04 | 0.40 | 0.084 | −0.353 | |
| Zlatar | 7.145 | 0.611 | 1.619 | 10.46 | 0.72 | 0.083 | −0.017 | |
| Tara | 6.625 | 0.606 | 1.576 | 10.06 | 0.23 | 0.012 | −0.037 | |
| Mean | 6.967 | 0.616 | 1.612 | 10.22 | 0.40 | 0.069 | −0.169 | 0.007 * |
| Population | Stara Planina | Kopaonik | Golija | Zlatar | Tara |
|---|---|---|---|---|---|
| Stara Planina | - | 0.009 * | 0.012 * | 0.019 * | 0.008 |
| Kopaonik | 0.016 * | - | 0.000 | 0.000 | 0.010 * |
| Golija | 0.020 * | 0.000 | - | 0.000 | 0.003 |
| Zlatar | 0.032 * | 0.000 | 0.000 | - | 0.008 * |
| Tara | 0.012 | 0.016 * | 0.002 | 0.012 * | - |
| Source of Variation | df | SS | Variance Component | Total Variance | F-Statistic | p Value |
|---|---|---|---|---|---|---|
| Among populations | 4 | 18.593 | 0.047 | 1 | 0.014 | 0.005 |
| Within populations | 145 | 470.167 | 3.243 | 99 | ||
| Total | 149 | 488.760 | 3.290 | 100 |
| Population | Pop ID | TPM Model | SMM Model | Mode Shift |
|---|---|---|---|---|
| Stara Planina | 1 | 0.625 | 0.062 | Normal |
| Kopaonik | 2 | 0.625 | 0.625 | Normal |
| Golija | 3 | 0.625 | 0.812 | Normal |
| Zlatar | 4 | 0.813 | 0.812 | Normal |
| Tara | 5 | 0.625 | 0.812 | Normal |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stojnić, S.; V. Avramidou, E.; Fussi, B.; Westergren, M.; Orlović, S.; Matović, B.; Trudić, B.; Kraigher, H.; A. Aravanopoulos, F.; Konnert, M. Assessment of Genetic Diversity and Population Genetic Structure of Norway Spruce (Picea abies (L.) Karsten) at Its Southern Lineage in Europe. Implications for Conservation of Forest Genetic Resources. Forests 2019, 10, 258. https://doi.org/10.3390/f10030258
Stojnić S, V. Avramidou E, Fussi B, Westergren M, Orlović S, Matović B, Trudić B, Kraigher H, A. Aravanopoulos F, Konnert M. Assessment of Genetic Diversity and Population Genetic Structure of Norway Spruce (Picea abies (L.) Karsten) at Its Southern Lineage in Europe. Implications for Conservation of Forest Genetic Resources. Forests. 2019; 10(3):258. https://doi.org/10.3390/f10030258
Chicago/Turabian StyleStojnić, Srđan, Evangelia V. Avramidou, Barbara Fussi, Marjana Westergren, Saša Orlović, Bratislav Matović, Branislav Trudić, Hojka Kraigher, Filippos A. Aravanopoulos, and Monika Konnert. 2019. "Assessment of Genetic Diversity and Population Genetic Structure of Norway Spruce (Picea abies (L.) Karsten) at Its Southern Lineage in Europe. Implications for Conservation of Forest Genetic Resources" Forests 10, no. 3: 258. https://doi.org/10.3390/f10030258