
Soil Microbial Community Composition and Diversity Analysis under Different Land Use Patterns in Taojia River Basin


 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
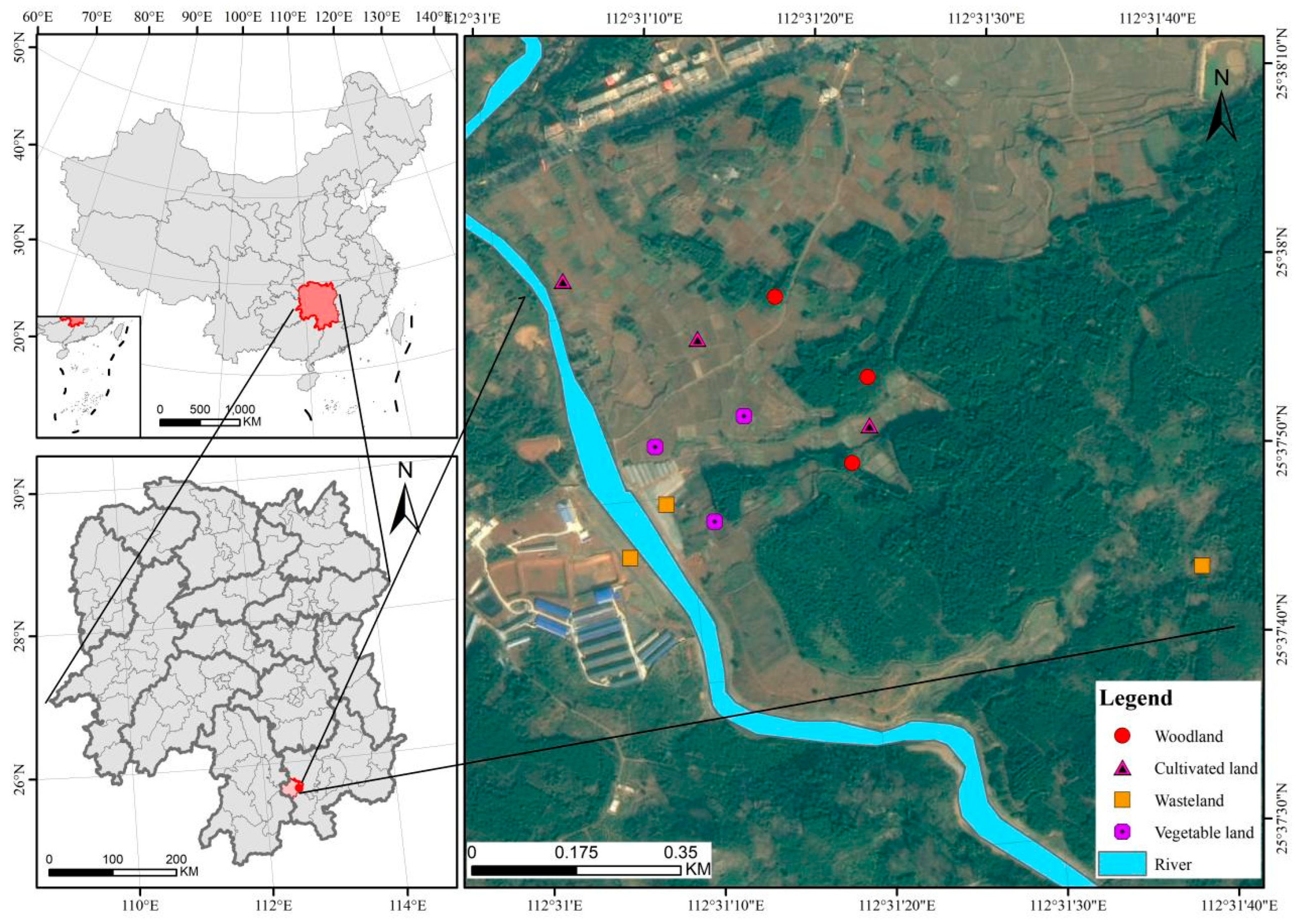
2.1. Location of the Study Area
2.2. Sampling
2.3. Determination of Soil Physicochemical Properties
2.4. Total Soil DNA Extraction, PCR Amplification, and Sequencing
2.5. Data Processing
3. Results
3.1. Analysis of Soil Physicochemical Properties under Different Land Use Patterns
3.2. Analysis of Soil Microbial Community Diversity under Different Land Use Patterns
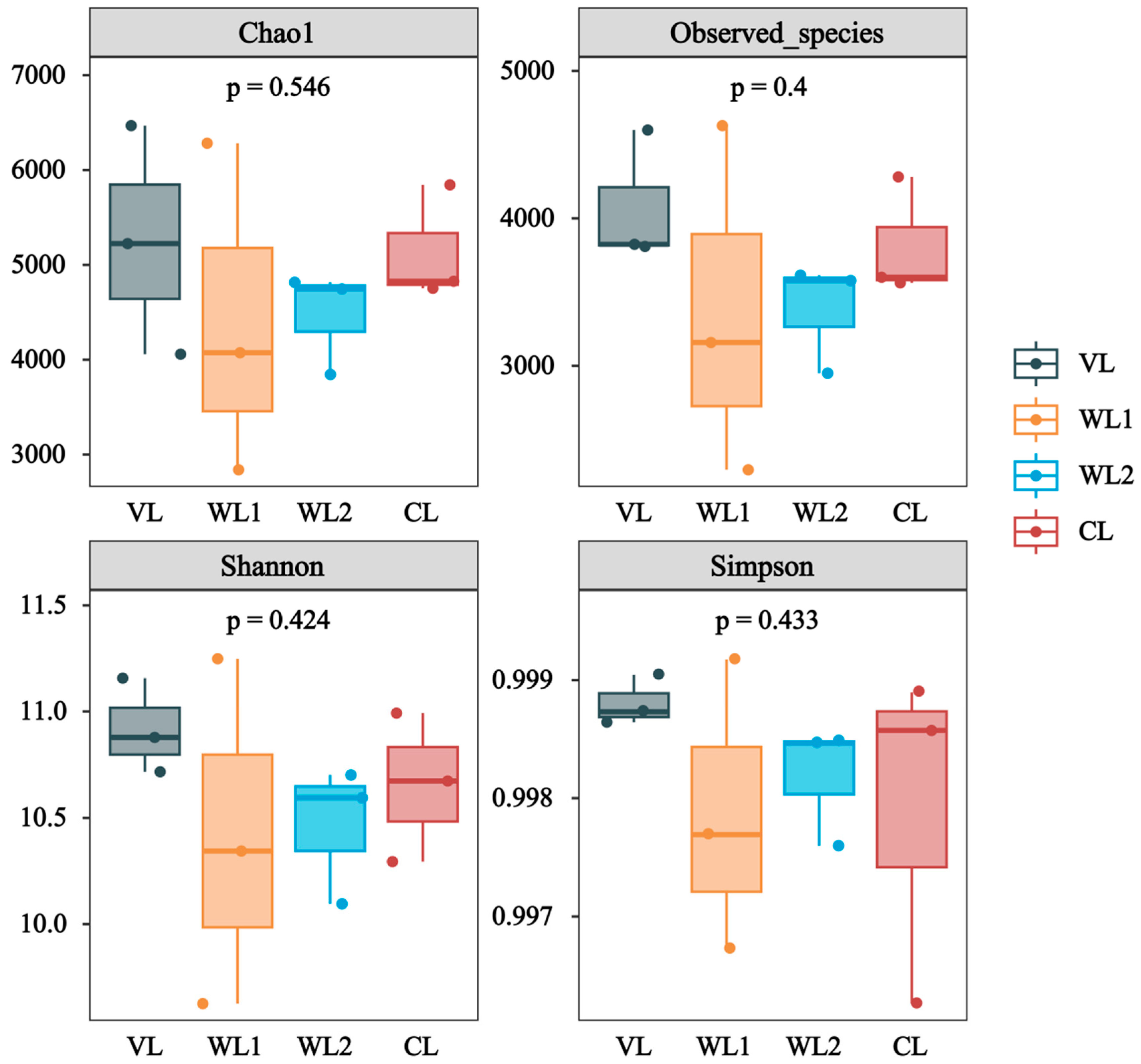
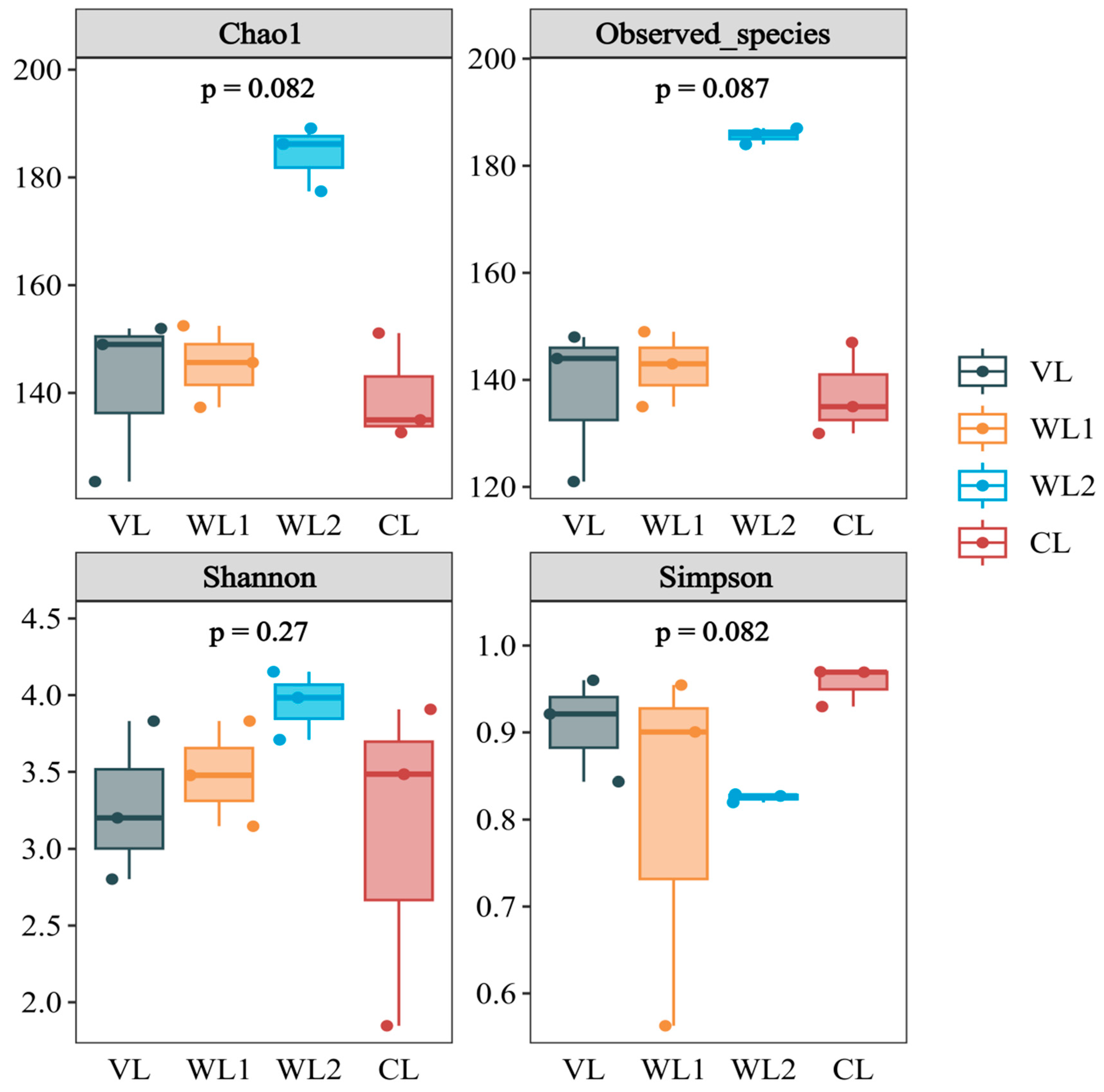
3.2.1. Analysis of Soil Bacterial Community Diversity
3.2.2. Analysis of Soil Fungal Community Diversity
3.3. Analysis of Soil Microbial Community Composition Characteristics under Different Land Use Patterns
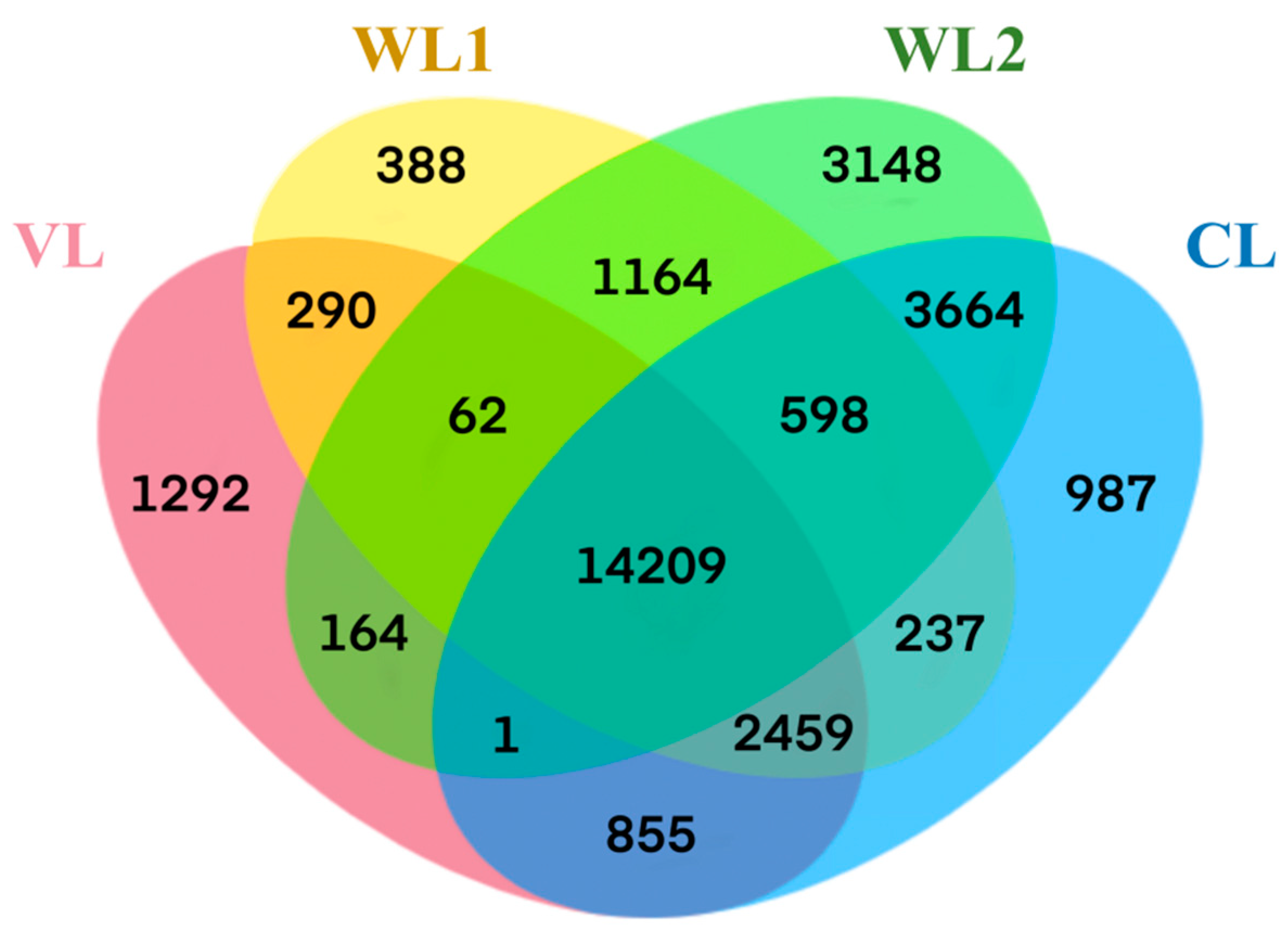
3.3.1. Analysis of Soil Bacterial Community Composition
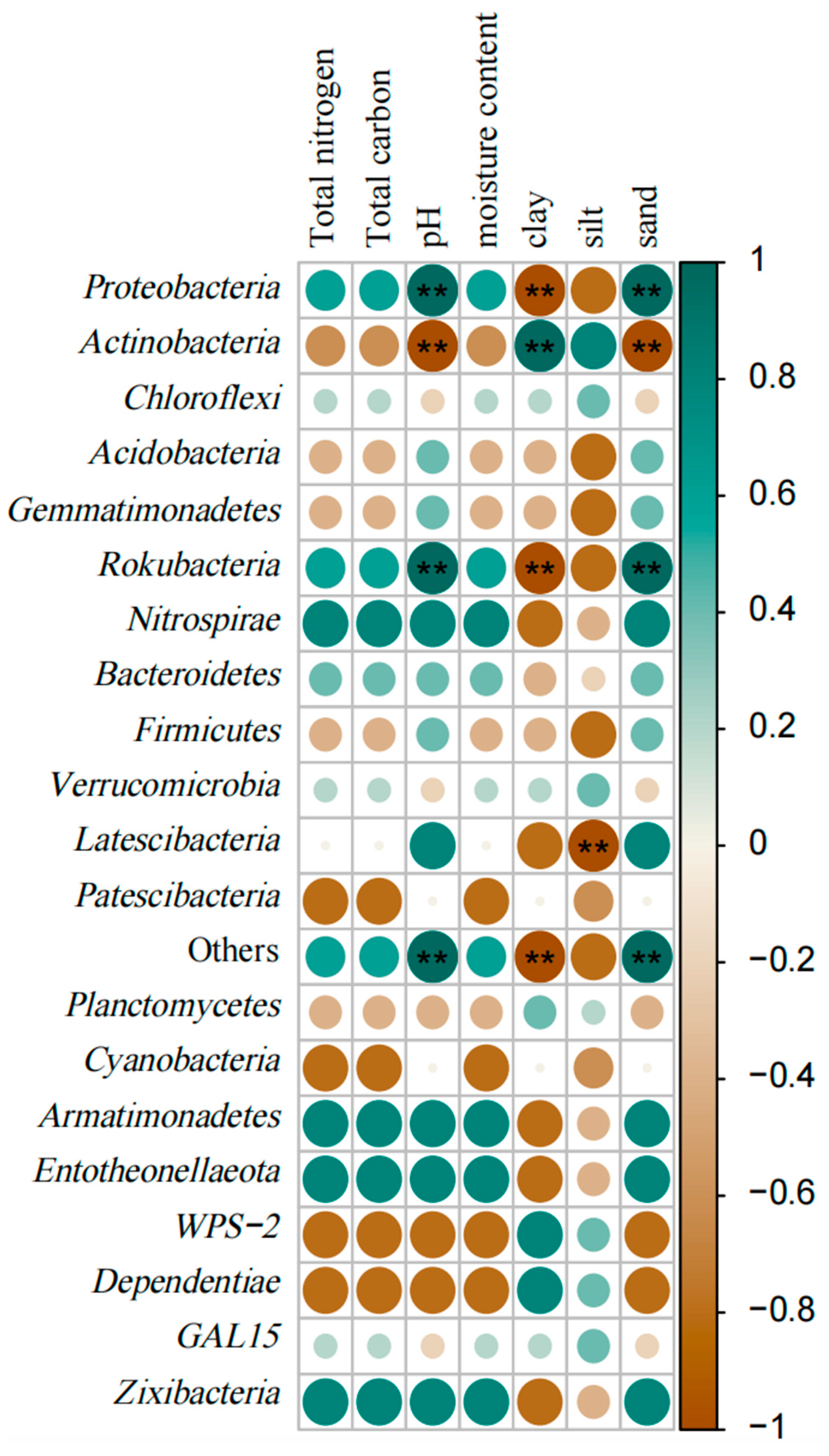
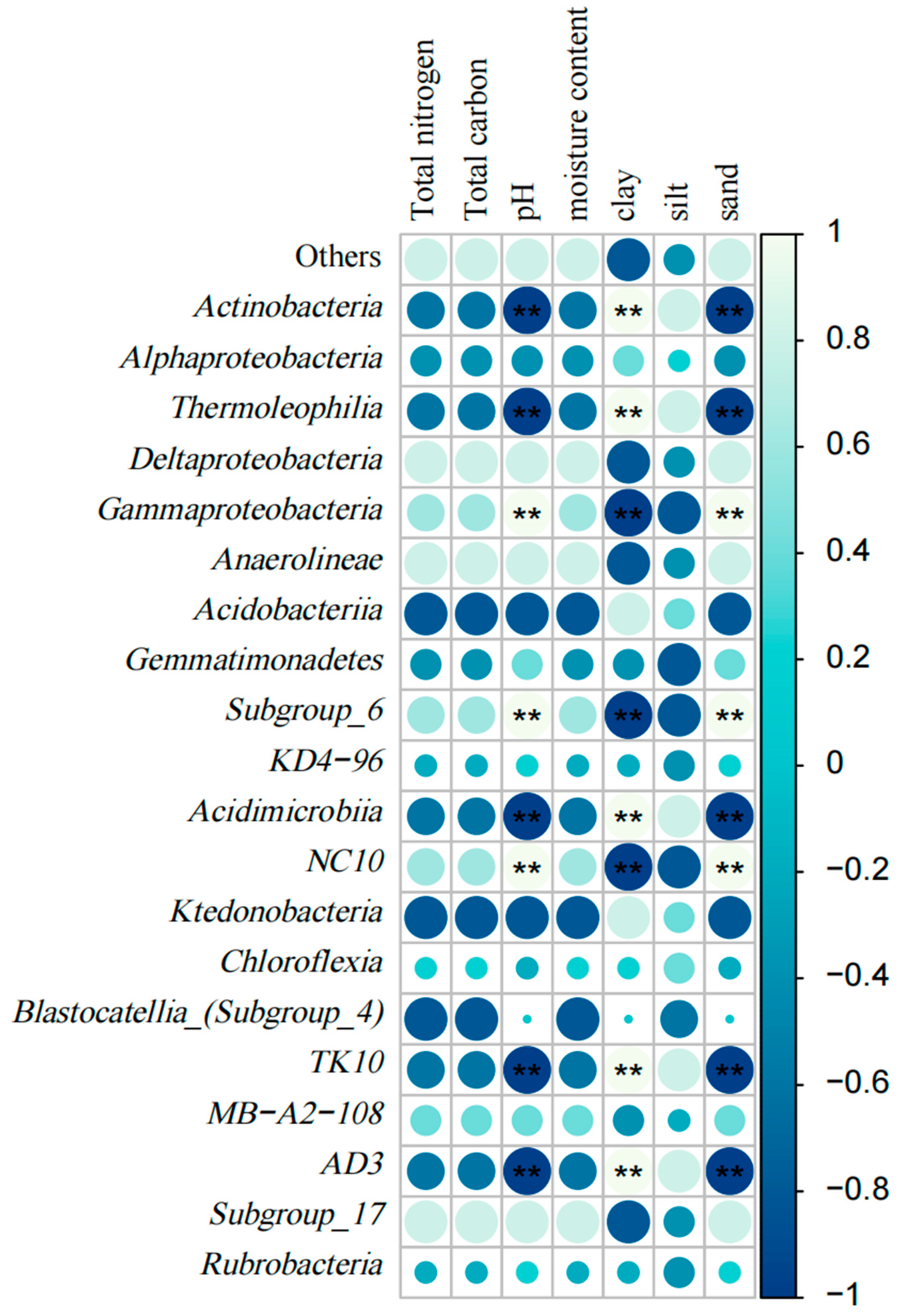
3.3.2. Correlation Analysis of Soil Bacterial Community Composition and Soil Physicochemical Properties
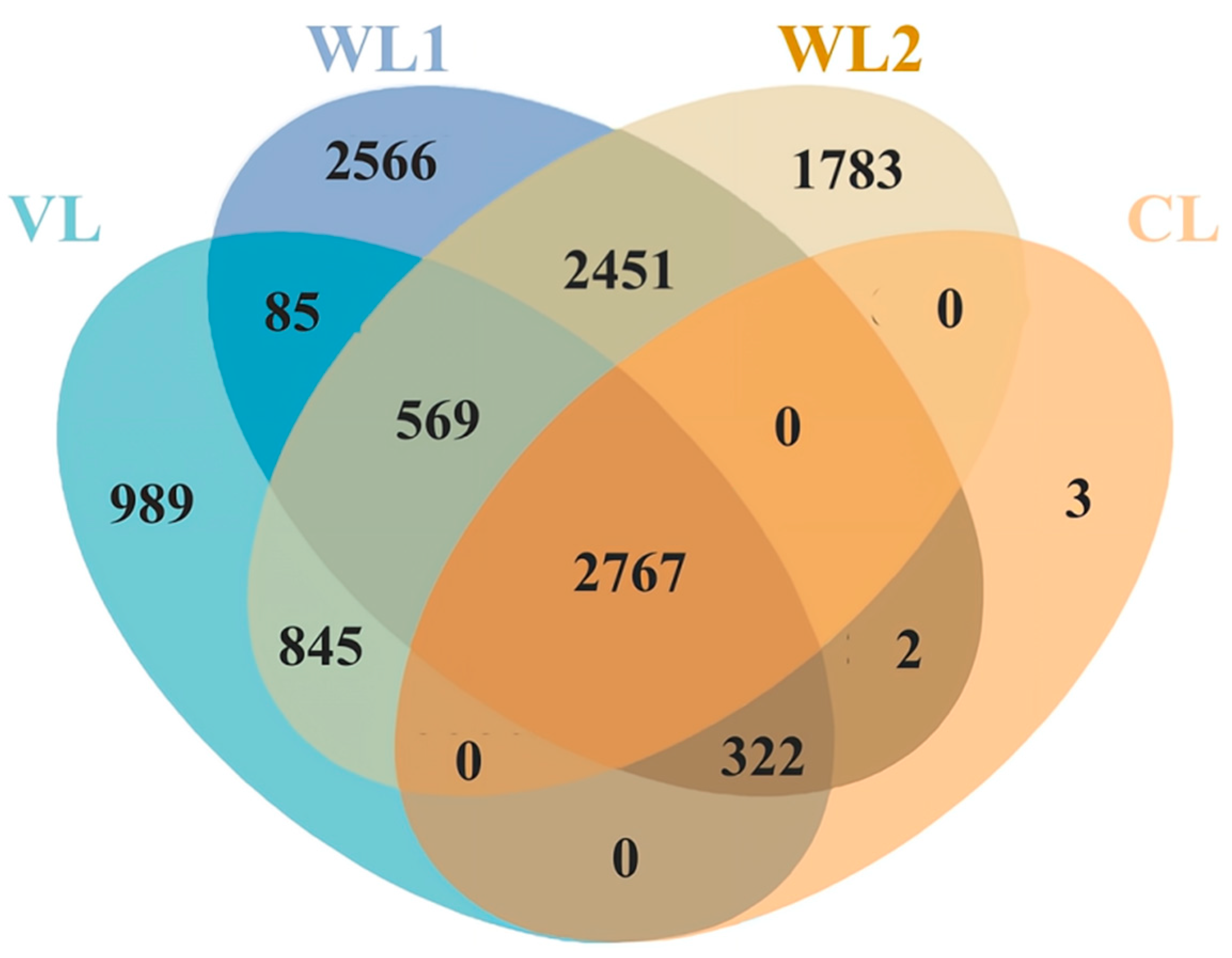
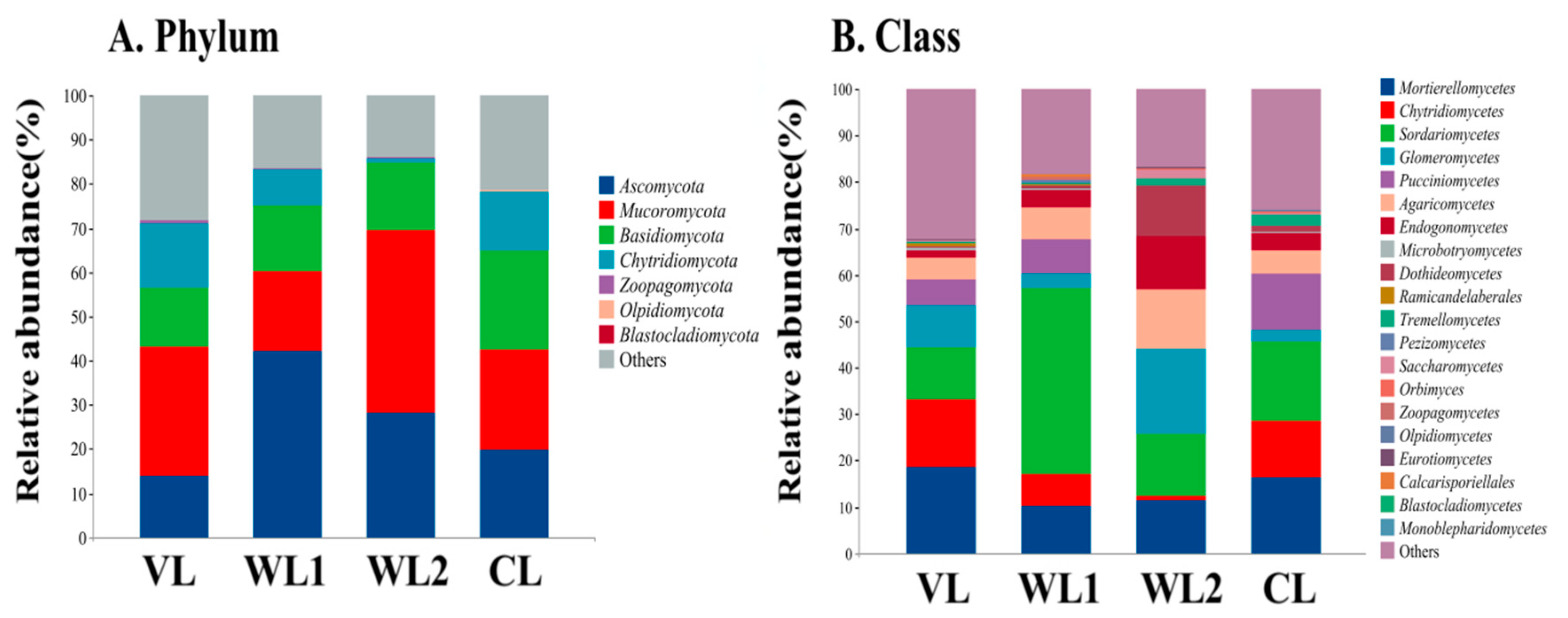
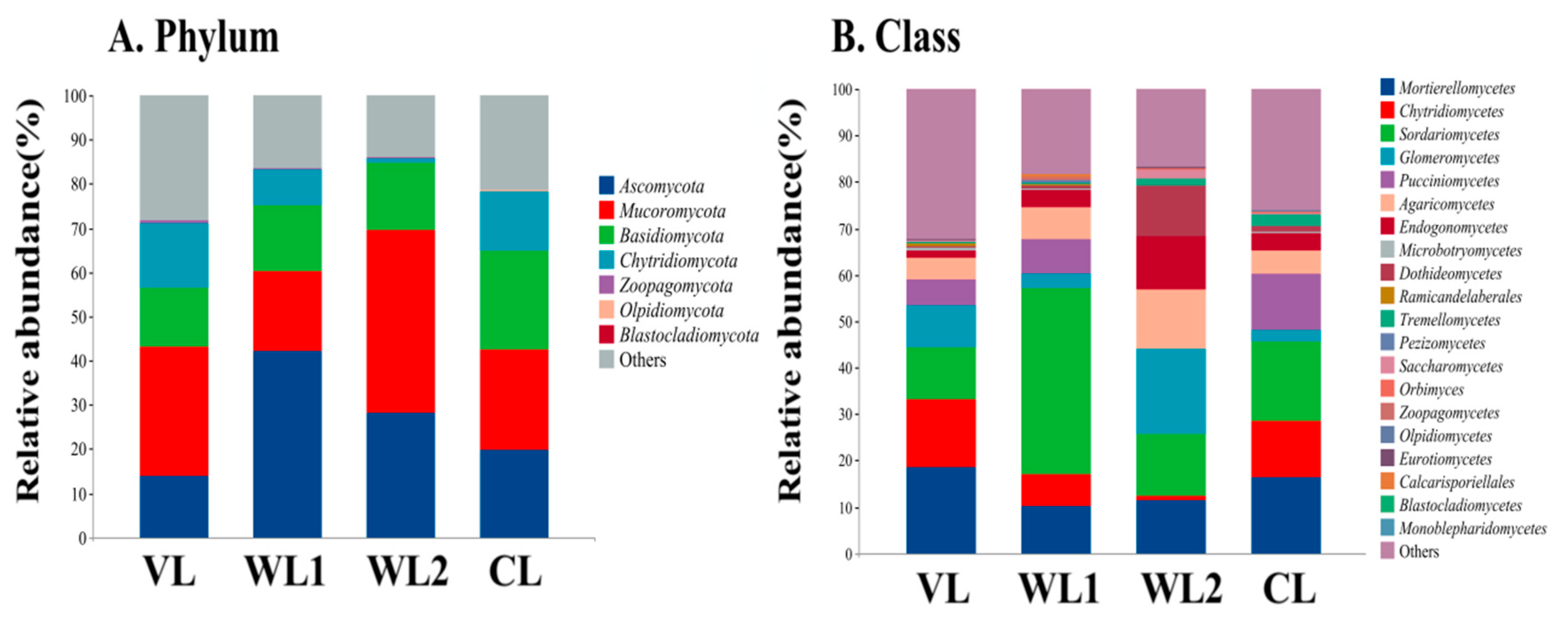
3.3.3. Analysis of Soil Fungal Community Composition
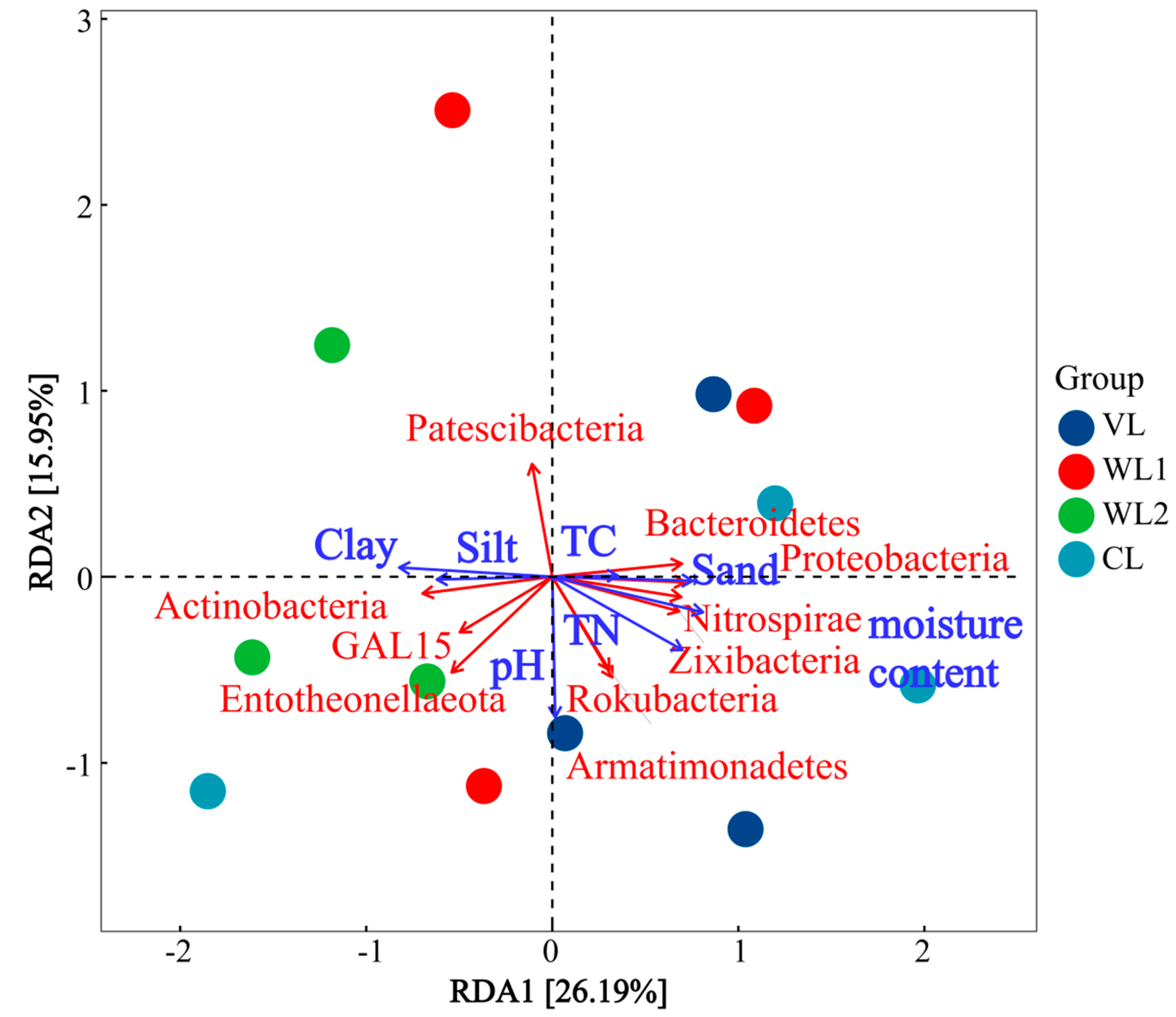
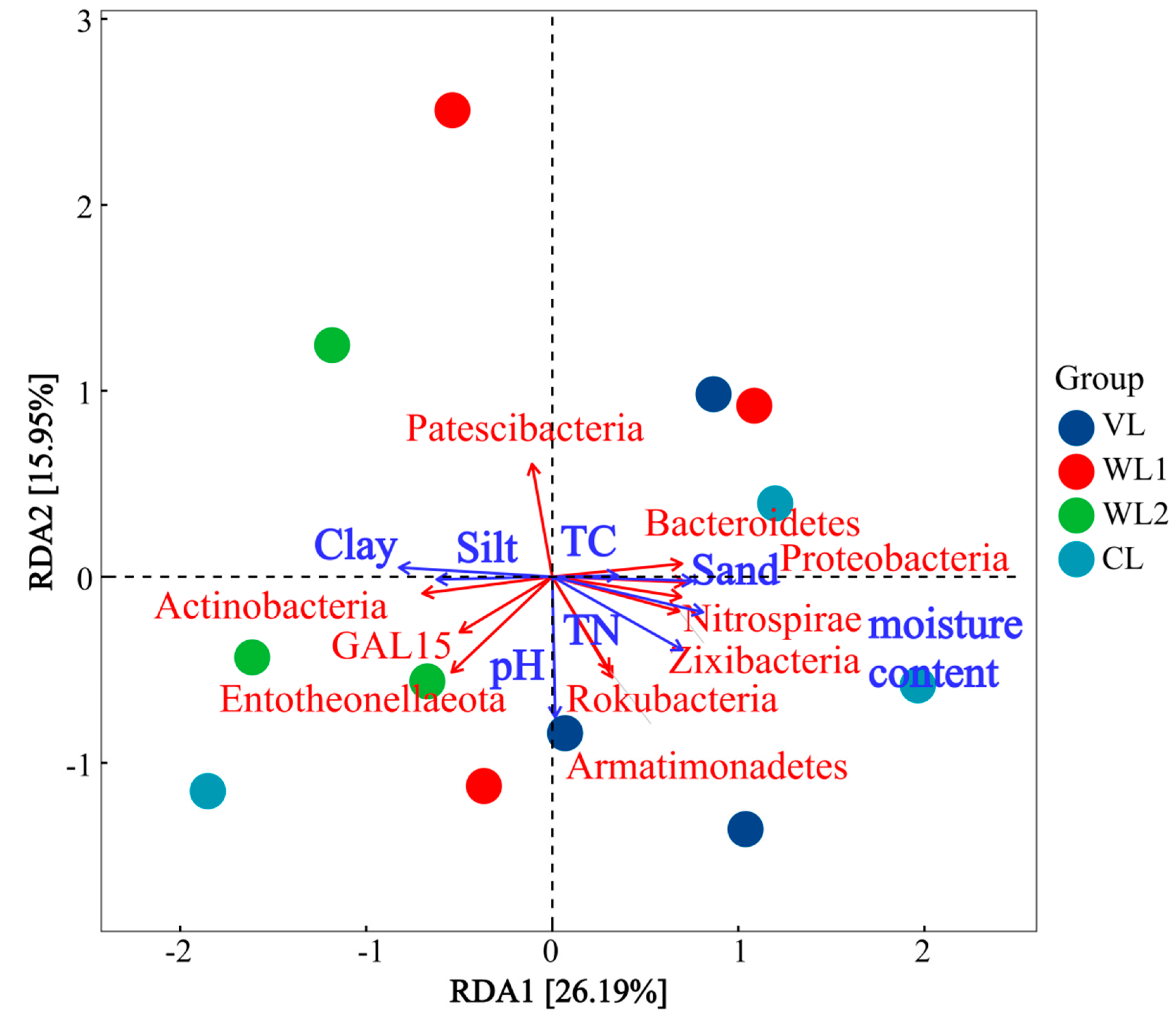
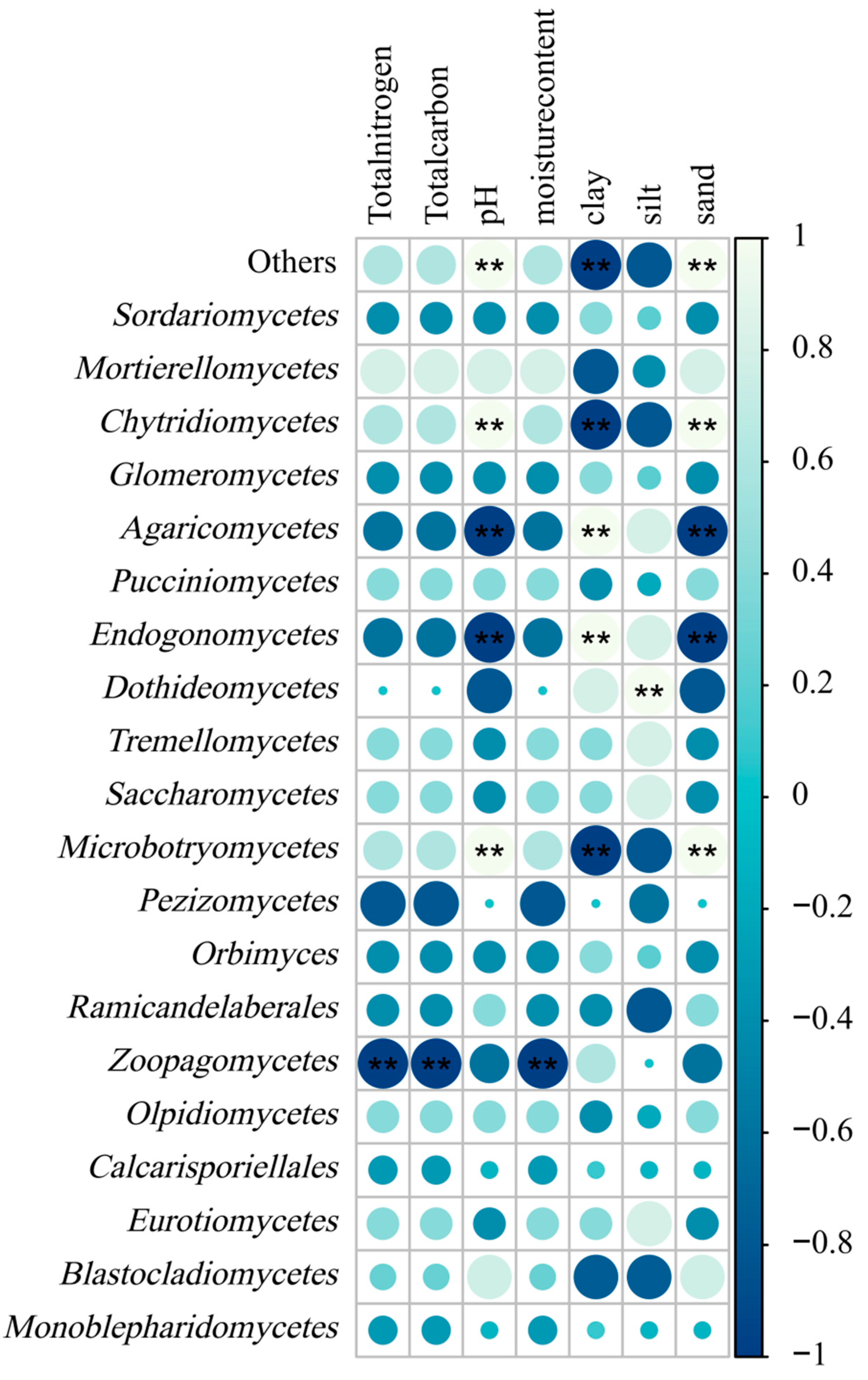
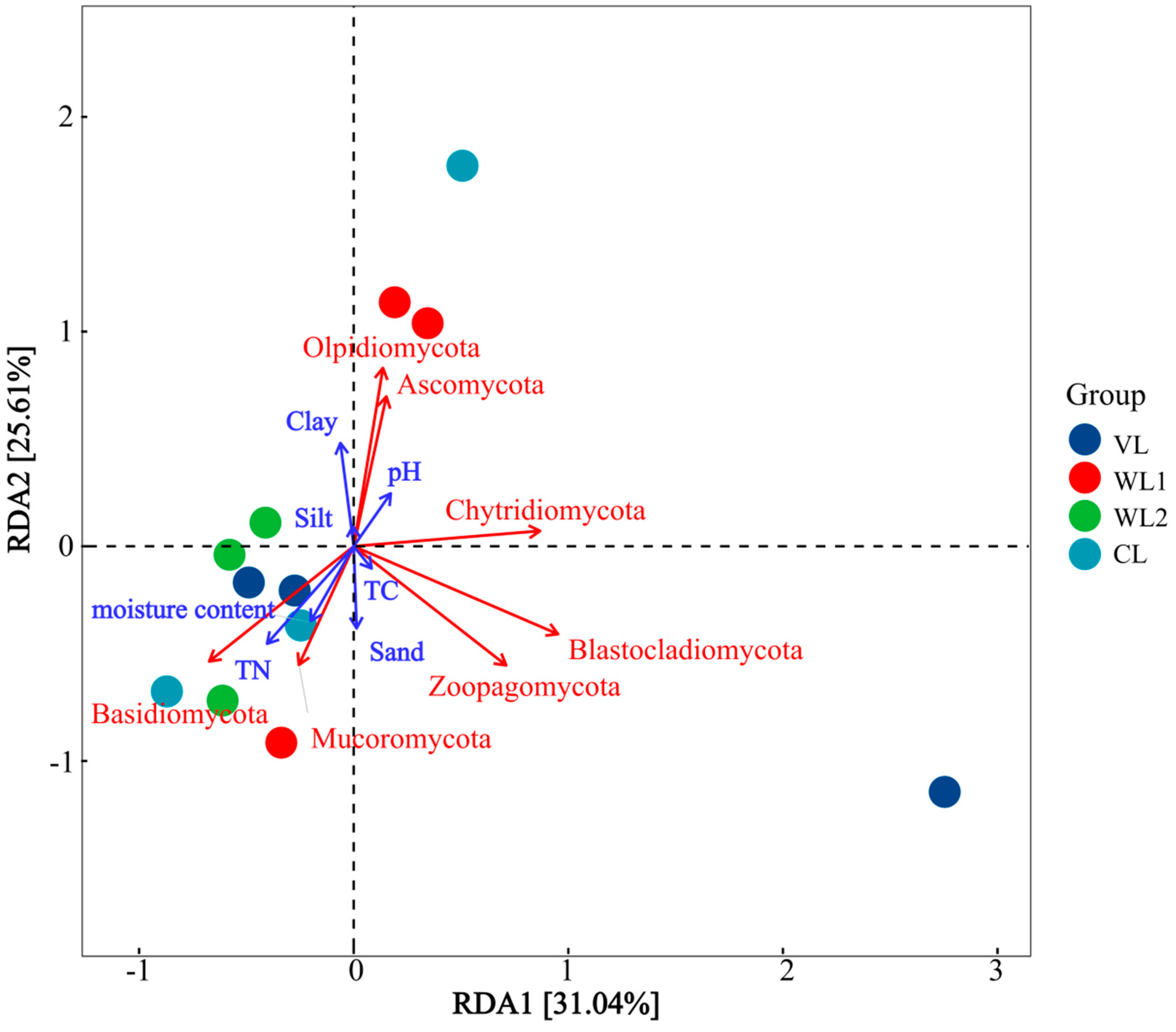
3.3.4. Correlation Analysis of Soil Fungal Community Composition and Soil Physicochemical Properties
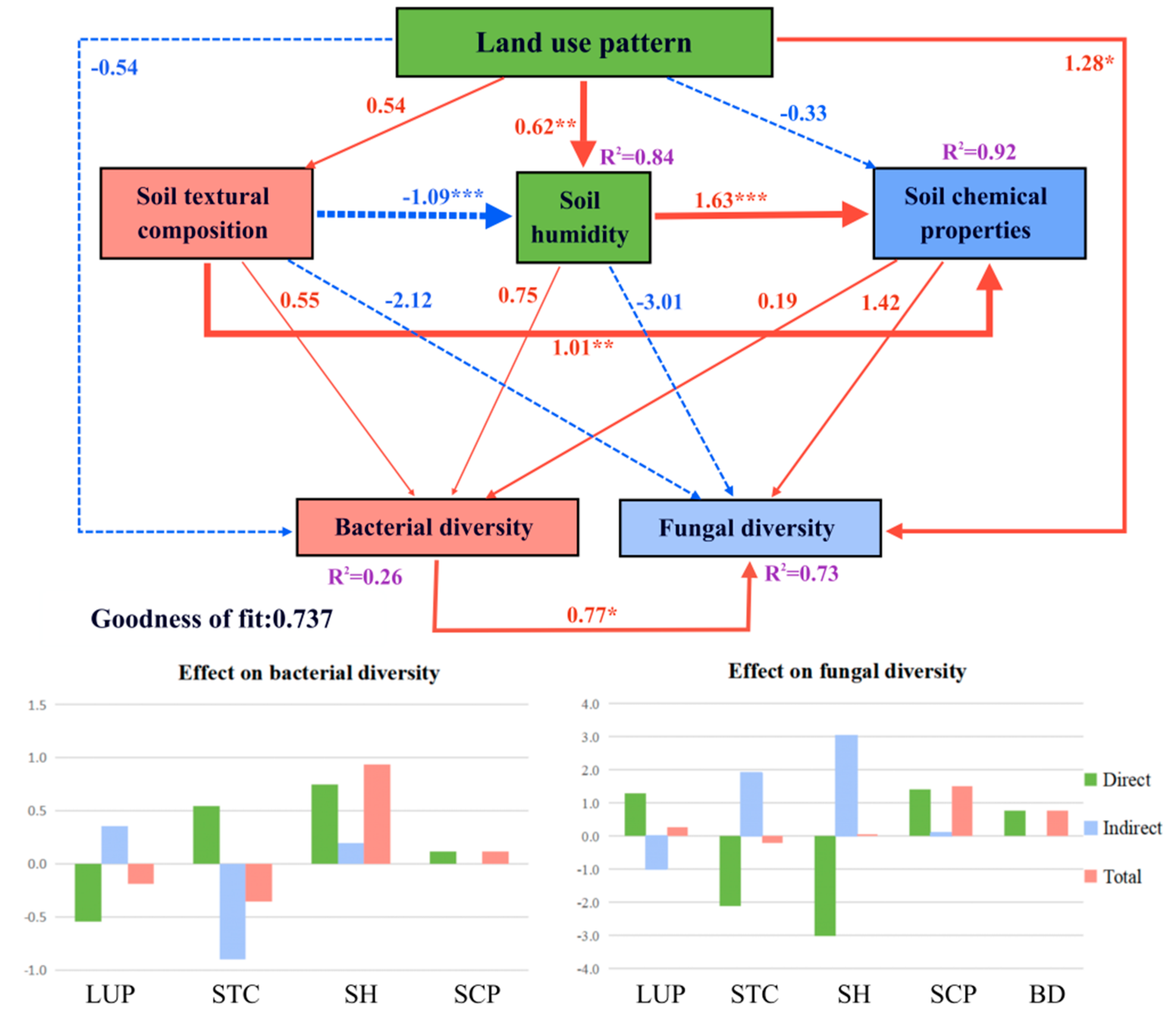
3.4. PLS-PM of Soil Bacterial and Fungal Community Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | VL | WL1 | WL2 | CL |
|---|---|---|---|---|
| Proteobacteria | 0.33 ± 0.01 a | 0.24 ± 0.01 b | 0.21 ± 0.03 b | 0.27 ± 0.08 ab |
| Chloroflexi | 0.17 ± 0.08 a | 0.19 ± 0.09 a | 0.18 ± 0.11 a | 0.21 ± 0.03 a |
| Actinobacteria | 0.16 ± 0.09 a | 0.24 ± 0.10 a | 0.39 ± 0.15 a | 0.18 ± 0.18 a |
| Acidobacteria | 0.16 ± 0.01 a | 0.16 ± 0.03 a | 0.12 ± 0.03 a | 0.15 ± 0.03 a |
| Gemmatimonadetes | 0.05 ± 0.01 a | 0.05 ± 0.01 a | 0.03 ± 0.01 a | 0.04 ± 0.01 a |
| Rokubacteria | 0.03 ± 0.01 a | 0.03 ± 0.02 a | 0.01 ± 0.01 a | 0.03 ± 0.00 a |
| Nitrospirae | 0.02 ± 0.01 ab | 0.02 ± 0.02 ab | 0.00 ± 0.00 b | 0.05 ± 0.03 a |
| Firmicutes | 0.01 ± 0.01 a | 0.02 ± 0.01 a | 0.01 ± 0.00 a | 0.01 ± 0.01 a |
| Bacteroidetes | 0.02 ± 0.01 a | 0.02 ± 0.01 a | 0.01 ± 0.00 a | 0.02 ± 0.01 a |
| Latescibacteria | 0.01 ± 0.01 a | 0.01 ± 0.00 ab | 0.00 ± 0.00 b | 0.01 ± 0.00 ab |
| Verrucomicrobia | 0.01 ± 0.01 a | 0.00 ± 0.00 a | 0.01 ± 0.00 a | 0.00 ± 0.00 a |
| GAL15 | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Cyanobacteria | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Planctomycetes | 0.01 ± 0.00 ab | 0.00 ± 0.00 ab | 0.01 ± 0.00 a | 0.00 ± 0.00 b |
| Patescibacteria | 0.01 ± 0.00 a | 0.01 ± 0.01 a | 0.01 ± 0.00 a | 0.00 ± 0.00 a |
| Entotheonellaeota | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.01 a |
| Armatimonadetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| WPS-2 | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.01 ± 0.01 a | 0.00 ± 0.00 a |
| Zixibacteria | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Dependentiae | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Taxon | VL | WL1 | WL2 | CL |
|---|---|---|---|---|
| Deltaproteobacteria | 0.11 ± 0.04 a | 0.08 ± 0.03 ab | 0.04 ± 0.02 b | 0.11 ± 0.03 a |
| Actinobacteria | 0.07 ± 0.02 b | 0.09 ± 0.01 b | 0.19 ± 0.05 a | 0.08 ± 0.05 b |
| Anaerolineae | 0.10 ± 0.08 a | 0.07 ± 0.08 a | 0.01 ± 0.01 a | 0.11 ± 0.05 a |
| Alphaproteobacteria | 0.10 ± 0.03 b | 0.09 ± 0.02 b | 0.14 ± 0.02 a | 0.08 ± 0.02 b |
| Thermoleophilia | 0.06 ± 0.06 a | 0.10 ± 0.07 a | 0.15 ± 0.10 a | 0.06 ± 0.08 a |
| Gammaproteobacteria | 0.12 ± 0.02 a | 0.07 ± 0.01 b | 0.03 ± 0.02 c | 0.08 ± 0.03 ab |
| Gemmatimonadetes | 0.04 ± 0.01 a | 0.05 ± 0.02 a | 0.03 ± 0.01 a | 0.04 ± 0.01 a |
| Acidobacteriia | 0.05 ± 0.02 a | 0.07 ± 0.07 a | 0.08 ± 0.04 a | 0.05 ± 0.03 a |
| NC10 | 0.03 ± 0.01 a | 0.03 ± 0.02 a | 0.01 ± 0.01 a | 0.03 ± 0.00 a |
| AD3 | 0.00 ± 0.00 a | 0.01 ± 0.01 a | 0.02 ± 0.03 a | 0.00 ± 0.00 a |
| Ktedonobacteria | 0.01 ± 0.00 a | 0.02 ± 0.02 a | 0.08 ± 0.09 a | 0.00 ± 0.00 a |
| Subgroup_6 | 0.05 ± 0.01 a | 0.04 ± 0.03 a | 0.02 ± 0.01 a | 0.04 ± 0.02 a |
| Acidimicrobiia | 0.02 ± 0.01 a | 0.03 ± 0.01 a | 0.04 ± 0.01 a | 0.03 ± 0.03 a |
| Chloroflexia | 0.01 ± 0.00 a | 0.02 ± 0.02 a | 0.02 ± 0.02 a | 0.03 ± 0.04 a |
| KD4-96 | 0.04 ± 0.01 a | 0.04 ± 0.02 a | 0.01 ± 0.01 a | 0.04 ± 0.02 a |
| Blastocatellia_(Subgroup_4) | 0.02 ± 0.01 a | 0.03 ± 0.03 a | 0.02 ± 0.02 a | 0.02 ± 0.00 a |
| TK10 | 0.01 ± 0.00 b | 0.02 ± 0.01 ab | 0.03 ± 0.02 a | 0.01 ± 0.00 b |
| MB-A2-108 | 0.01 ± 0.01 a | 0.01 ± 0.01 a | 0.01 ± 0.01 a | 0.01 ± 0.02 a |
| Rubrobacteria | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Subgroup_17 | 0.01 ± 0.00 ab | 0.00 ± 0.00 ab | 0.00 ± 0.00 b | 0.01 ± 0.00 a |
| Taxon | VL | WL1 | WL2 | CL |
|---|---|---|---|---|
| Ascomycota | 0.14 ± 0.10 a | 0.42 ± 0.29 a | 0.28 ± 0.17 a | 0.20 ± 0.20 a |
| Mucoromycota | 0.29 ± 0.16 a | 0.18 ± 0.09 a | 0.41 ± 0.10 a | 0.23 ± 0.12 a |
| Basidiomycota | 0.13 ± 0.08 a | 0.15 ± 0.13 a | 0.15 ± 0.02 a | 0.22 ± 0.05 a |
| Chytridiomycota | 0.15 ± 0.15 a | 0.08 ± 0.04 a | 0.01 ± 0.00 a | 0.13 ± 0.09 a |
| Zoopagomycota | 0.00 ± 0.01 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Olpidiomycota | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Blastocladiomycota | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Taxon | VL | WL1 | WL2 | CL |
|---|---|---|---|---|
| Sordariomycetes | 0.11 ± 0.10 a | 0.40 ± 0.30 a | 0.13 ± 0.04 a | 0.17 ± 0.18 a |
| Dothideomycetes | 0.00 ± 0.01 a | 0.01 ± 0.01 a | 0.11 ± 0.18 a | 0.01 ± 0.01 a |
| Glomeromycetes | 0.09 ± 0.08 ab | 0.03 ± 0.03 ab | 0.18 ± 0.13 a | 0.03 ± 0.02 b |
| Agaricomycetes | 0.05 ± 0.03 b | 0.07 ± 0.00 b | 0.13 ± 0.02 a | 0.05 ± 0.03 b |
| Mortierellomycetes | 0.19 ± 0.21 a | 0.10 ± 0.06 a | 0.11 ± 0.07 a | 0.16 ± 0.07 a |
| Chytridiomycetes | 0.15 ± 0.15 a | 0.07 ± 0.03 a | 0.01 ± 0.00 a | 0.12 ± 0.08 a |
| Endogonomycetes | 0.02 ± 0.01 a | 0.04 ± 0.06 a | 0.11 ± 0.17 a | 0.04 ± 0.04 a |
| Pucciniomycetes | 0.06 ± 0.06 a | 0.07 ± 0.13 a | 0.00 ± 0.00 a | 0.12 ± 0.12 a |
| Tremellomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.01 ± 0.01 a | 0.03 ± 0.04 a |
| Calcarisporiellales | 0.00 ± 0.00 a | 0.00 ± 0.01 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Eurotiomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0,00 a |
| Saccharomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.02 ± 0.02 a | 0.00 ± 0.00 a |
| Orbimyces | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Microbotryomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.01 a |
| Ramicandelaberales | 0.00 ± 0.01 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Pezizomycetes | 0.00 ± 0.00 a | 0.01 ± 0.01 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Olpidiomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Zoopagomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Blastocladiomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
| Monoblepharidomycetes | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a | 0.00 ± 0.00 a |
References
- Yao, H.; He, Z.; Wilson, M.J.; Campbell, C.D. Microbial Biomass and Community Structure in a Sequence of Soils with Increasing Fertility and Changing Land Use. Microb. Ecol. 2000, 40, 223–237. [Google Scholar] [CrossRef]
- Yang, X.; Leng, Y.; Zhou, Z.; Shang, H.; Ni, K.; Ma, L.; Yi, X.; Cai, Y.; Ji, L.; Ruan, J.; et al. Ecological Management Model for the Improvement of Soil Fertility through the Regulation of Rare Microbial Taxa in Tea (Camellia sinensis L.) Plantation Soils. J. Environ. Manag. 2022, 308, 114595. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Rabbi, S.M.F.; Zhang, Y.; Young, I.M.; Jones, A.R.; Dennis, P.G.; Menzies, N.W.; Kopittke, P.M.; Dalal, R.C. Soil Organic Carbon Is Significantly Associated with the Pore Geometry, Microbial Diversity and Enzyme Activity of the Macro-Aggregates under Different Land Uses. Sci. Total Environ. 2021, 778, 146286. [Google Scholar] [CrossRef] [PubMed]
- Rillig, M.C.; Aguilar-Trigueros, C.A.; Bergmann, J.; Verbruggen, E.; Veresoglou, S.D.; Lehmann, A. Plant Root and Mycorrhizal Fungal Traits for Understanding Soil Aggregation. New Phytol. 2015, 205, 1385–1388. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Li, Z.; Wu, F.; Tang, M. Microenvironment and Microbial Community in the Rhizosphere of Dioecious Populus Cathayana at Chaka Salt Lake. J. Soils Sediments 2019, 19, 2740–2751. [Google Scholar] [CrossRef]
- Rillig, M.C.; Mummey, D.L. Mycorrhizas and Soil Structure. New Phytol. 2006, 171, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Vlček, V.; Pohanka, M. Glomalin—An Interesting Protein Part of the Soil Organic Matter. Soil Water Res. 2020, 15, 67–74. [Google Scholar] [CrossRef]
- Wang, M.; Qu, L.; Ma, K.; Yuan, X. Soil Microbial Properties under Different Vegetation Types on Mountain Han. Sci. China Life Sci. 2013, 56, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Du, H.; Peng, W.; Liu, Y.; Fang, Z.; Song, T. Functional Diversity of the Soil Culturable Microbial Community in Eucalyptus Plantations of Different Ages in Guangxi, South China. Forests 2019, 10, 1083. [Google Scholar] [CrossRef]
- Dangi, S.R.; Stahl, P.D.; Wick, A.F.; Ingram, L.J.; Buyer, J.S. Soil Microbial Community Recovery in Reclaimed Soils on a Surface Coal Mine Site. Soil Sci. Soc. Am. J. 2012, 76, 915–924. [Google Scholar] [CrossRef]
- Yan, B.; Li, J.; Xiao, N.; Qi, Y.; Fu, G.; Liu, G.; Qiao, M. Urban-Development-Induced Changes in the Diversity and Composition of the Soil Bacterial Community in Beijing. Sci. Rep. 2016, 6, 38811. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wei, Y.; Yin, Y.; Ding, H.; Zhu, W.; Zhou, Y. The Effect of Intercropping Mulberry (Morus alba L.) with Peanut (Arachis hypogaea L.), on the Soil Rhizosphere Microbial Community. Forests 2022, 13, 1757. [Google Scholar] [CrossRef]
- Ji, H.; Wei, H.; Wang, R.; Zhang, J.; Liu, Z.; Abdellah, Y.A.Y.; Ren, X.; Shan, X.; Zhong, J.; He, Z. Heterogeneity and Its Drivers of Microbial Communities and Diversity in Six Typical Soils under Two Different Land Uses in Tropical and Subtropical Southern China. Appl. Soil Ecol. 2022, 179, 104555. [Google Scholar] [CrossRef]
- Zhou, J.; He, Z.; Yang, Y.; Deng, Y.; Tringe, S.G.; Alvarez-Cohen, L. High-Throughput Metagenomic Technologies for Complex Microbial Community Analysis: Open and Closed Formats. Mbio 2015, 6, e02288-14. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, C.; Long, M. Variation of Soil Nutrients and Bacterial Community Diversity of Different Land Utilization Types in Yangtze River Basin, Chongqing Municipality. PeerJ 2020, 8, e9386. [Google Scholar] [CrossRef]
- Li, J.; Pokharel, P.; Liu, G.; Chen, J. Reclamation of Desert Land to Different Land-Use Types Changes Soil Bacterial Community Composition in a Desert-Oasis Ecotone. Land. Degrad. Dev. 2021, 32, 1389–1399. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, W.; Zou, Y.; Dong, H.; Yu, F.; Wang, H.; Zong, C. Effects of Land Use Conversion on the Soil Microbial Community Composition and Functionality in the Urban Wetlands of North-Eastern China. Forests 2022, 13, 1148. [Google Scholar] [CrossRef]
- He, X.; Su, Y.; Liang, Y.; Chen, X.; Zhu, H.; Wang, K. Land Reclamation and Short-Term Cultivation Change Soil Microbial Communities and Bacterial Metabolic Profiles. J. Sci. Food Agric. 2012, 92, 1103–1111. [Google Scholar] [CrossRef]
- Wang, X.; Yang, J.; Xie, X.; Chen, X.; Pu, L.; Zhang, X. Soil Microbial Succession with Soil Development since Costal Reclamation. Catena 2020, 187, 104393. [Google Scholar] [CrossRef]
- Yuan, C.; Liang, S.; Wu, X.; Farooq, T.H.; Liu, T.; Hu, Y.; Wang, G.; Wang, J.; Yan, W. Land Use Changes Influence the Soil Enzymatic Activity and Nutrient Status in the Polluted Taojia River Basin in Sub-Tropical China. Int. J. Environ. Res. Public Health 2022, 19, 13999. [Google Scholar] [CrossRef]
- Cao, H.; Wang, Z.; Liao, X.; Li, Y.; Zhu, Y. Research on the Regional Environmental Impact and Risk Assessment Affected by Mineral Resource Development: A Case Study of the Taojia River Watershed in Hunan. Front. Ecol. Evol. 2022, 10, 918446. [Google Scholar] [CrossRef]
- Nachtergaele, F. Soil Taxonomy—A Basic System of Soil Classification for Making and Interpreting Soil Surveys. Geoderma 2001, 3, 336–337. [Google Scholar] [CrossRef]
- Chen, L.C.; Guan, X.; Li, H.M.; Wang, Q.K.; Zhang, W.D.; Yang, Q.P.; Wang, S.L. Spatiotemporal Patterns of Carbon Storage in Forest Ecosystems in Hunan Province, China. For. Ecol. Manag. 2019, 432, 656–666. [Google Scholar] [CrossRef]
- Weaver, B.; Wuensch, K.L. SPSS and SAS Programs for Comparing Pearson Correlations and OLS Regression Coefficients. Behav. Res. Methods 2013, 45, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, H.; Yi, H. Impact of Fertilizer on Crop Yield and C:N:P Stoichiometry in Arid and Semi-Arid Soil. Int. J. Environ. Res. Public Health 2021, 18, 4341. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Yang, L.; Harbo, L.S.; Yan, X.; Chen, J.; Zhao, C.; Xiao, Y.; Liu, H.; Wang, S.; Miao, Y.; et al. Effects of Land Use on Soil Microbial Community Structure and Diversity in the Yellow River Floodplain. J. Plant Ecol. 2023, 16, rtac075. [Google Scholar] [CrossRef]
- Ge, C.; Xue, D.; Yao, H. Microbial Biomass, Community Diversity, and Enzyme Activities in Response to Urea Application in Tea Orchard Soils. Commun. Soil Sci. Plant Anal. 2010, 41, 797–810. [Google Scholar] [CrossRef]
- Li, Y.T.; Rouland, C.; Benedetti, M.; Li, F.; Pando, A.; Lavelle, P.; Dai, J. Microbial Biomass, Enzyme and Mineralization Activity in Relation to Soil Organic C, N and P Turnover Influenced by Acid Metal Stress. Soil Biol. Biochem. 2009, 41, 969–977. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, A.; Qin, X.; Yu, H.; Ji, X.; He, S.; Zong, Y.; Wang, J.; Tang, J. Effects of Intercropping Pandanus Amaryllifolius on Soil Properties and Microbial Community Composition in Areca Catechu Plantations. Forests 2022, 13, 1814. [Google Scholar] [CrossRef]
- Miśkowiec, P.; Olech, Z. Searching for the Correlation between the Activity of Urease and the Content of Nickel in the Soil Samples: The Role of Metal Speciation. J. Soil Sci. Plant Nutr. 2020, 20, 1904–1911. [Google Scholar] [CrossRef]
- Weller, D.M. Pseudomonas Biocontrol Agents of Soilborne Pathogens: Looking Back over 30 Years. Phytopathology 2007, 97, 250–256. [Google Scholar] [CrossRef] [PubMed]
- May, L.A.; Smiley, B.; Schmidt, M.G. Comparative Denaturing Gradient Gel Electrophoresis Analysis of Fungal Communities Associated with Whole Plant Corn Silage. Can. J. Microbiol 2001, 47, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Lemanowicz, J. Dynamics of Phosphorus Content and the Activity of Phosphatase in Forest Soil in the Sustained Nitrogen Compounds Emissions Zone. Environ. Sci. Pollut Res. 2018, 25, 33773–33782. [Google Scholar] [CrossRef]
- Fu, D.; Wu, X.; Qiu, Q.; Duan, C.; Jones, D.L. Seasonal Variations in Soil Microbial Communities under Different Land Restoration Types in a Subtropical Mountains Region, Southwest China. Appl. Soil Ecol. 2020, 153, 103634. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences From Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classififier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microb. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Liu, K.; Xue, Y.Q.; Zhu, L.; Xu, F.; Zhu, Z.; Zhang, T. Effect of Different Land Use Types on the Diversity of Soil Bacterial Community in the Coastal Zone of Jialing River. Environ. Sci. 2021, 43, 1620–1629. [Google Scholar]
- Yang, W.; Cai, X.; Wang, Y.; Diao, L.; Xia, L.; An, S.; Luo, Y.; Cheng, X. Increased Soil Bacterial Abundance but Decreased Bacterial Diversity and Shifted Bacterial Community Composition Following Secondary Succession of Old-Field. Forests 2022, 13, 1628. [Google Scholar] [CrossRef]
- Kuramae, E.E.; Yergeau, E.; Wong, L.C.; Pijl, A.S.; Veen, J.A.; Kowalchuk, G.A. Soil Characteristics More Strongly Influence Soil Bacterial Communities than Land-Use Type. FEMS Microbiol. Ecol. 2012, 79, 12–24. [Google Scholar] [CrossRef]
- Gong, J.; Hou, W.; Liu, J.; Malik, K.; Kong, X.; Wang, L.; Chen, X.; Tang, M.; Zhu, R.; Cheng, C.; et al. Effects of Different Land Use Types and Soil Depths on Soil Mineral Elements, Soil Enzyme Activity, and Fungal Community in Karst Area of Southwest China. Int. J. Environ. Res. Public Health 2022, 19, 3120. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, S.K.; Gao, Q.Z.; Liu, S.L.; Zhou, H.K.; Ganjurjav, H.; Wang, X. Climate Change and Human Activities Altered the Diversity and Composition of Soil Microbial Community in Alpine Grasslands of the QinghaiTibetan Plateau. Sci. Total Environ. 2016, 562, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.F.; Bai, Y.; Xiang, Q.J.; Yu, X.M.; Zhao, K.; Zhang, X.P.; Li, C.; Liu, S.; Chen, Q. Degradation Shaped Bacterial and Archaeal Communities with Predictable Taxa and Their Association Patterns in Zoige Wetland at Tibet Plateau. Sci. Rep. 2018, 8, 3884. [Google Scholar] [CrossRef]
- Hemmat-Jou, M.H.; Safari-Sinegani, A.A.; Che, R.; Mirzaie-Asl, A.; Tahmourespour, A.; Tahmasbian, I. Toxic Trace Element Resistance Genes and Systems Identified Using the Shotgun Metagenomics Approach in an Iranian Mine Soil. Environ. Sci. Pollut Res. 2021, 28, 4845–4856. [Google Scholar] [CrossRef]
- Fernández-Cadena, J.C.; Ruíz-Fernández, P.S.; Fernández-Ronquillo, T.E.; Díez, B.; Trefault, N.; Andrade, S.; De la Iglesia, R. Detection of Sentinel Bacteria in Mangrove Sediments Contaminated with Heavy Metals. Mar. Pollut. Bull 2020, 150, 110701. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Chen, W.; Wang, J.; Du, N.; Li, Q.; Wei, G. Soil Microbiomes with Distinct Assemblies through Vertical Soil Profiles Drive the Cycling of Multiple Nutrients in Reforested Ecosystems. Microbiome 2018, 6, 146. [Google Scholar] [CrossRef]
- Zhang, R.; Rong, L.; Zhang, L. Soil Nutrient Variability Mediates the Effects of Erosion on Soil Microbial Communities: Results from a Modified Topsoil Removal Method in an Agricultural Field in Yunnan Plateau, China. Environ. Sci. Pollut. Res. 2022, 29, 3659–3671. [Google Scholar] [CrossRef]
- Yan, N.; Marschner, P.; Cao, W.; Zuo, C.; Qin, W. Influence of Salinity and Water Content on Soil Microorganisms. Int. Soil Water Conse 2015, 3, 316–323. [Google Scholar] [CrossRef]
- Zeng, J.; Shen, J.P.; Wang, J.T.; Hu, H.W.; Zhang, C.J.; Bai, R.; Zhang, L.-M.; He, J.-Z. Impacts of Projected Climate Warming and Wetting on Soil Microbial Communities in Alpine Grassland Ecosystems of the Tibetan Plateau. Microb. Ecol. 2018, 75, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, D.G.; Jiang, Z.H.; Sun, P.; Xiao, H.L.; Wu, Y.X.; Chen, J. Changes in the Soil Microbial Communities of Alpine Steppe at Qinghai-Tibetan Plateau under Different Degradation Levels. Sci. Total Environ. 2019, 651, 2281–2291. [Google Scholar] [CrossRef]
- Liu, J.; Engel, B.A.; Wang, Y.; Zhang, G.; Zhang, Z.; Zhang, M. Multi-Scale Analysis of Hydrological Connectivity and Plant Response in the Yellow River Delta. Sci. Total Environ. 2020, 702, 134889. [Google Scholar] [CrossRef]
- Ulea, E.; Lipsa, F.D.; Balau, A.M.; Filipov, F.; Morari, E.C. Diversity of Soil Bacteria as Indicator of Soil Pollution in Moldavia Region, Romania. Environ. Eng. Manag. J. 2017, 16, 879–889. [Google Scholar]
- Zhang, G.; Jia, J.; Zhao, Q.; Wang, W.; Wang, D.; Bai, J. Seasonality and Assembly of Soil Microbial Communities in Coastal Salt Marshes Invaded by a Perennial Grass. J. Environ. Manag. 2023, 331, 117247. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zheng, L.; Zhou, Y.; Sang, W.; Zhao, J.; Liu, L.; Li, C.; Xiao, C. Changes in Soil Microbial Community Structure and Function Following Degradation in a Temperate Grassland. J. Plant Ecol. 2021, 14, 384–397. [Google Scholar] [CrossRef]
- Zeng, X.M.; Feng, J.; Chen, J.; Delgado-Baquerizo, M.; Zhang, Q.; Zhou, X.Q.; Yuan, Y.; Feng, S.; Zhang, K.; Liu, Y.R.; et al. Microbial Assemblies Associated with Temperature Sensitivity of Soil Respiration along an Altitudinal Gradient. Sci. Total Environ. 2022, 820, 153257. [Google Scholar] [CrossRef] [PubMed]



| Samples | VL | WL1 | WL2 | CL | F | p |
|---|---|---|---|---|---|---|
| Total nitrogen (g·kg−1) | 0.14 ± 0.03 a | 0.11 ± 0.08 a | 0.11 ± 0.01 a | 0.17 ± 0.04 a | 0.908 | 0.479 |
| Total carbon (g·kg−1) | 16.56 ± 1.21 ab | 13.51 ± 0.45 c | 14.45 ± 1.44 bc | 17.99 ± 2.14 a | 5.929 | 0.020 |
| pH | 6.30 ± 0.29 a | 5.57 ± 0.99 a | 5.47 ± 0.14 a | 6.24 ± 0.76 a | 1.373 | 0.319 |
| moisture content (%) | 30.25 ± 4.63 a | 20.98 ± 3.02 b | 21.27 ± 2.02 b | 30.80 ± 6.04 a | 4.988 | 0.031 |
| Clay (%) | 4.92 ± 2.29 b | 10.81 ± 0.05 a | 12.23 ± 1.07 a | 9.29 ± 4.44 ab | 3.694 | 0.070 |
| Silt (%) | 30.90 ± 4.47 b | 35.33 ± 1.54 ab | 38.81 ± 0.85 a | 36.15 ± 2.37 ab | 3.439 | 0.081 |
| Sand (%) | 64.17 ± 6.71 a | 52.00 ± 0.36 b | 48.96 ± 1.93 b | 54.57 ± 6.77 ab | 4.399 | 0.049 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.; Yuan, C.; Chen, P.; Rong, Z.; Peng, T.; Farooq, T.H.; Wang, G.; Yan, W.; Wang, J. Soil Microbial Community Composition and Diversity Analysis under Different Land Use Patterns in Taojia River Basin. Forests 2023, 14, 1004. https://doi.org/10.3390/f14051004
He Z, Yuan C, Chen P, Rong Z, Peng T, Farooq TH, Wang G, Yan W, Wang J. Soil Microbial Community Composition and Diversity Analysis under Different Land Use Patterns in Taojia River Basin. Forests. 2023; 14(5):1004. https://doi.org/10.3390/f14051004
Chicago/Turabian StyleHe, Zhe, Chenglin Yuan, Peirou Chen, Ziqiang Rong, Ting Peng, Taimoor Hassan Farooq, Guangjun Wang, Wende Yan, and Jun Wang. 2023. "Soil Microbial Community Composition and Diversity Analysis under Different Land Use Patterns in Taojia River Basin" Forests 14, no. 5: 1004. https://doi.org/10.3390/f14051004
APA StyleHe, Z., Yuan, C., Chen, P., Rong, Z., Peng, T., Farooq, T. H., Wang, G., Yan, W., & Wang, J. (2023). Soil Microbial Community Composition and Diversity Analysis under Different Land Use Patterns in Taojia River Basin. Forests, 14(5), 1004. https://doi.org/10.3390/f14051004