
De Novo Assembly and Characterization of Bud, Leaf and Flowers Transcriptome from Juglans Regia L. for the Identification and Characterization of New EST-SSRs

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collections, DNA Extraction and RNA Extraction
2.2. RNA-seq Library Preparation for Transcriptome Sequencing
2.3. Transcriptome Assembling and Gene Annotation
2.4. Discovery of EST-SSRs, Primer Design, Amplification Conditions, and Marker Validation
2.5. Population Genetics Data Analysis
2.6. Data Deposit
3. Results
3.1. Sequence Assembly
3.2. Gene Annotation of J. regia Transcriptomes
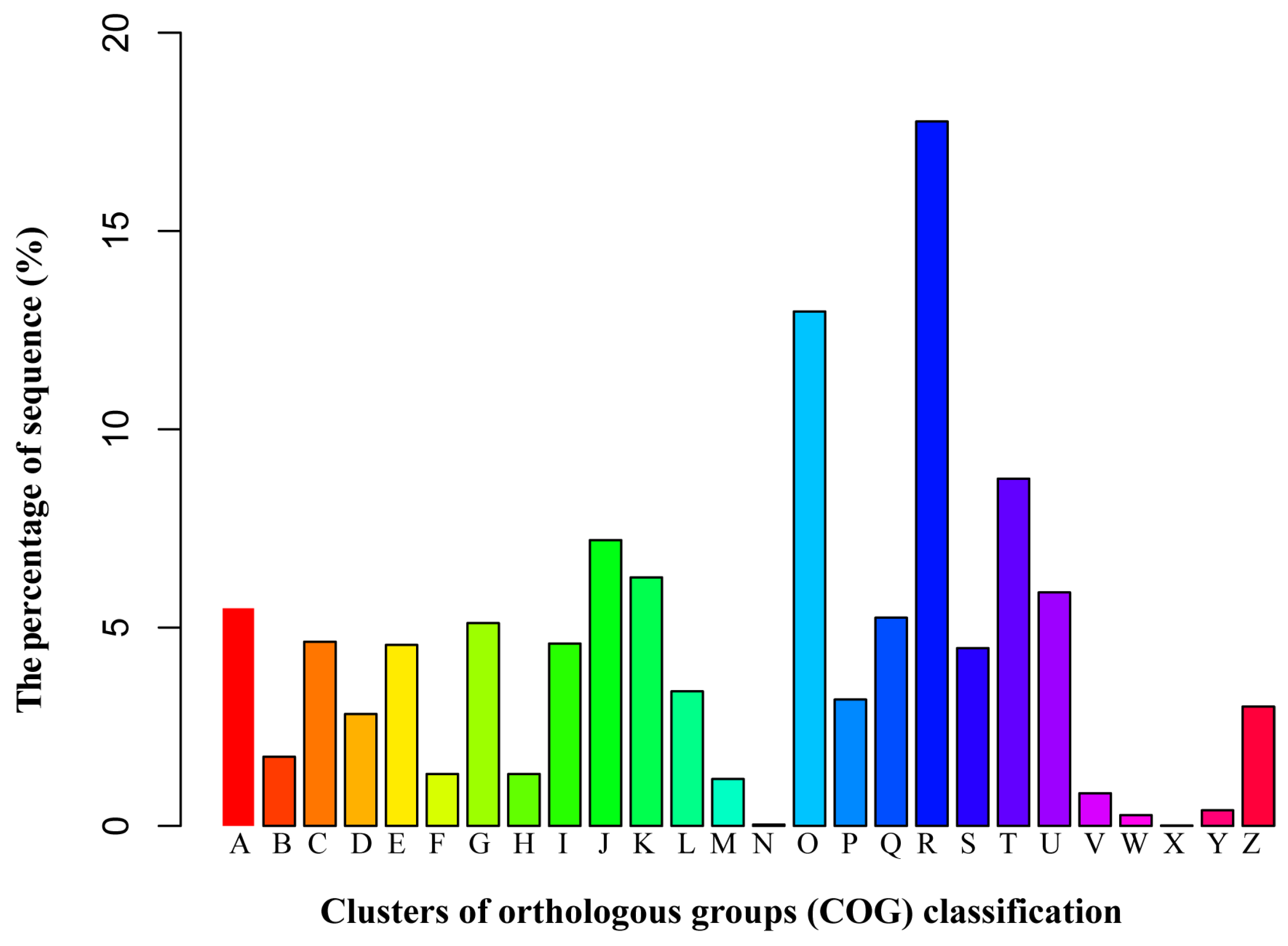
3.3. Functional Classification by the Orthologous Groups (COG)
3.4. Functional Classification by the KEGG Pathway
3.5. Distribution of the SSRs in Transcriptomes, SSR Primer Screening and Verification
3.6. Assessment of Genetic Diversity and Population Structure of J. regia in China Using 39 EST-SSRs
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Woeste, K.; Michler, C. Genomic and breeding resources. In Wild Crop Relatives; Chittaranjan, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Kodad, O.; Sindic, M. Kernel quality in a local walnut (Juglans regia) population grown under different ecological conditions in Morocco. Nucis Newsl. 2014, 16, 27–31. [Google Scholar]
- Pollegioni, P.; Woeste, K.E.; Chiocchini, F.; Del Lungo, S.; Olimpieri, I.; Tortolano, V.; Clark, J.; Hemery, E.G.; Mapelli, S.; Malvolti, M.E. Ancient humans influenced the current spatial genetic structure of common walnut populations in Asia. PLoS ONE 2015, 10, e0135980. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.L.; Labuckas, D.O.; Lamarque, A.L.; Maestri, D.M. Walnut (Juglans regia L.): Genetic resources, chemistry, by-products. J. Sci. Food Agric. 2010, 90, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Rorabaugh, J.M.; Singh, A.P.; Sherrell, I.M.; Freeman, M.R.; Vorsa, N.; Fitschen, P.; Malone, C.; Maher, M.A.; Wilson, T. English and Black Walnut phenolic antioxidant activity in vitro and following human nut consumption. Food Nutr. Sci. 2011, 2, 193–200. [Google Scholar] [CrossRef]
- Vinson, J.A.; Cai, Y. Nuts, especially walnuts, have both antioxidant quantity and efficacy and exhibit significant potential health benefits. Food Funct. 2012, 3, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organisation; FAOSTAT Data; FAO: Rome, Italy, 2008.
- Topçu, H.; Ikhsan, A.S.; Sütyemez, M.; Çoban, N.; Güney, M.; Kafkas, S. Development of 185 polymorphic simple sequence repeat (SSR) markers from walnut (Juglans regia L.). Sci. Hortic. 2015, 194, 160–167. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, A.; Wang, X.; Yu, J.; Zhang, H.; Gao, J.; Deng, X. Development of Juglans regia SSR markers by data mining of the EST database. Plant Mol. Biol. Rep. 2010, 28, 646–653. [Google Scholar] [CrossRef]
- Wei, W.; Qi, X.; Wang, L.; Zhang, Y.; Hua, W.; Li, D.; Lv, H.; Zhang, X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Sigmund, R.; Börner, A.; Korzun, V.; Stein, N.; Sorrells, M.E.; Langridge, P.; Graner, A. Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci. 2005, 168, 195–202. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Han, J.W.; Jin, Q.; Wang, Y.; Pang, X.M.; Li, Y.Y. Development and characterization of new microsatellites for walnut (Juglans regia). Genet. Mol. Res. 2013, 12, 4723–4734. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Pembleton, L.W.; Cogan, N.O.; Savin, K.W.; Leonforte, T.; Paull, J.; Materne, M.; Forster, J.W. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genom. 2012, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Izzah, N.K.; Lee, J.; Jayakodi, M.; Perumal, S.; Jin, M.; Park, B.S.; Ahn, K.; Yang, T.J. Transcriptome sequencing of two parental lines of cabbage (Brassica oleracea L. var. capitata L.) and construction of an EST-based genetic map. BMC Genom. 2014, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.A.; Swain, M.T.; Hegarty, M.J.; Chernukin, I.; Lowe, M.; Allison, G.G.; Skøt, L. De novo assembly of red clover transcriptome based on RNA–Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Qiao, G.; Jiang, J.; Yang, H.; Xie, L.; Xie, J.; Zhuo, R. Transcriptome sequencing and de novo analysis for ma bamboo (Dendrocalamus latiflorus Munro) using the Illumina platform. PLoS ONE 2012, 7, e46766. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Dinkins, R.D.; Hunt, A.G. De novo transcriptome assembly and dynamic spatial gene expression analysis in red clover. Plant Gen. 2016, 9. [Google Scholar] [CrossRef]
- Wang, X.W.; Luan, J.B.; Li, J.M.; Bao, Y.Y.; Zhang, C.X.; Liu, S.S. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genom. 2010, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Wei, C.; Yang, H.; Zhang, L.; Chen, Q.; Deng, W.; Zhang, J.; Fang, C.; Ho, C.; Wan, X. Transcriptomic and phytochemical analysis of the biosynthesis of characteristic constituents in tea (Camellia sinensis) compared with oil tea (Camellia oleifera). BMC Plant Biol. 2015, 15, 190. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Liu, Z.X.; Chen, X.; Zhang, T.; Zhou, H.J.; Hu, Y.H.; Zhao, P. Identification, development, and application of 12 polymorphic EST-SSR markers for an endemic Chinese walnut (Juglans cathayensis L.) using next-generation sequencing technology. Biochem. Syst. Ecol. 2015, 60, 74–80. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, F.; Tan, H.W.; Li, M.Y.; Xu, Z.S.; Tan, G.F.; Xiong, A.S. De novo transcriptome assembly, gene annotation, marker development, and miRNA potential target genes validation under abiotic stresses in Oenanthe javanica. Mol. Genet. Genom. 2015, 290, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhang, T.; Gao, X.X.; Wang, Y.; Zhang, Q.; Zhou, H.J.; Zhao, G.F.; Wang, M.L.; Zhao, P. De novo assembly and characterization of the leaf, bud, and fruit transcriptome from the vulnerable tree Juglans mandshurica for the development of 20 new microsatellite markers using Illumina sequencing. Mol. Genet. Genom. 2016, 291, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.; Doyle, J.L. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Zhao, P.; Woeste, K.E. DNA markers identify hybrids between butternut (Juglans cinerea L.) and Japanese walnut (Juglans ailantifolia Carr.). Tree Genet. Genomes 2011, 7, 511–533. [Google Scholar] [CrossRef]
- Novogene Bioinformatics Technology Co. Available online: http://www.novogene.cn (accessed on 28 August 2016).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Chen, Z. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J. WEGO: A web tool for plotting GO annotations. Nucleic. Acids. Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Zhang, J.; Tian, X.; Wu, S.; Zhang, Q.; Zhang, J.; Dang, Z.; Pei, X.W. De novo assembly of the desert tree Haloxylon ammodendron (C. A. Mey.) based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 1111. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.O.; Donovan, C.; Phan, I.; et al. The SWISSPROT protein knowledgebase and its supplement TrEMBL. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-through put functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2013, 106, 411–422. [Google Scholar]
- Rozen, S.; Skaletsky, H.J. Primer3. Code. 1998. Available online: http://www-genome.wi.mit.edu/genome_software/other/primer3.html (accessed on 28 August 2016).
- Peakall, R.O.D.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Bohonak, A.J. IBD (isolation by distance): A program for analyses of isolation by distance. J. Hered. 2002, 93, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.L.; Bohonak, A.J.; Kelley, S.T. Isolation by distance, web service. BMC Genet. 2005, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Li, D.; Deng, Z.; Qin, B.; Liu, X.; Men, Z. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom. 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Najafi, F.; Mardi, M.; Fakheri, B.; Pirseyedi, S.M.; Mehdinejad, N.; Farsi, M. Isolation and characterization of novel microsatellite markers in walnut (Juglans regia L.). Am. J. Plant Sci. 2014, 5, 409. [Google Scholar] [CrossRef]
- Du, Q.; Gong, C.; Pan, W.; Zhang, D. Development and application of microsatellites in candidate genes related to wood properties in the Chinese white poplar (Populus tomentosa Carr.). DNA Res. 2012, 20, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Xiao, Y.; Liu, Z.; Luo, Y.; Mason, A.S.; Fan, H.; Yang, Y.; Zhao, S.; Peng, M. Development of gene-based simple sequence repeat markers for association analysis in Cocos nucifera. Mol. Breed. 2014, 34, 525–535. [Google Scholar] [CrossRef]
- Hou, X.J.; Liu, S.R.; Khan, M.R.G.; Hu, C.G.; Zhang, J.Z. Genome-wide identification, classification expression profiling and SSR marker development of the MADS-box gene family in Citrus. Plant Mol. Biol. Rep. 2014, 32, 28–41. [Google Scholar] [CrossRef]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bodénès, C.; Chancerel, E.; Gailing, O.; Vendramin, G.G.; Bagnoli, F.; Durand, J.; Goicoechea, P.G.; Villani, F.; Mattioni, C.; Koelewijn, H.P.; et al. Comparative mapping in the Fagaceae and beyond with EST-SSRs. BMC Plant Biol. 2012, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Barbara, T.; Palma-Silva, C.; Paggi, G.M.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Fan, L.; Liu, Q.Z.; Song, Y.; Wei, S.W.; Zhang, S.L.; Wu, J. A novel set of EST-derived SSR markers for pear and cross-species transferability in Rosaceae. Plant Mol. Biol. Rep. 2014, 32, 290–302. [Google Scholar] [CrossRef]
- Chen, L.; Ma, Q.; Chen, Y.; Wang, B.; Pei, D. Identification of major walnut cultivars grown in China based on nut phenotypes and SSR markers. Sci. Hortic. 2014, 168, 240–248. [Google Scholar] [CrossRef]
- Li, G.T.; Ai, C.X.; Zhang, L.S.; Wei, H.R.; Liu, Q.Z. ISSR analysis of genetic diversity among seedling walnut (Juglans spp.) populations. J. Plant Genet. Resour. 2011, 12, 640–645. (In Chinese) [Google Scholar]
- Ning, D.; Ma, Q.; Zhang, Y.; Wang, H.; Liu, B.; Pei, D. FISH-AFLP analysis of genetic diversity on walnut cultivars in Yunnan Province. For. Res. 2011, 24, 189–193. (In Chinese) [Google Scholar]
- Hu, Y.H.; Dang, M.; Zhang, T.; Luo, G.C.; Xia, H.L.; Zhou, H.J.; Hu, D.F.; He, L.; Ma, Z.H.; Zhao, P. Genetic diversity and evolutionary relationship of Juglans regia Wild and domesticated populations in Qinling Mountains based on nrDNA ITS sequences. Scientia Silvae Sinicae 2014, 50, 47–55. (In Chinese) [Google Scholar]
- Wang, H.; Pei, D.; Gu, R.S.; Wang, B.Q. Genetic diversity and structure of walnut populations in central and southwestern China revealed by microsatellite markers. J. Am. Soc. Hortic. Sci. 2008, 133, 197–203. [Google Scholar]
- Gunn, B.F.; Aradhya, M.; Salick, J.M.; Miller, A.J.; Yongping, Y.; Lin, L.; Xian, H. Genetic variation in walnuts (Juglans regia and J. Sigillata; Juglandaceae): Species distinctions, human impacts, and the conservation of agrobiodiversity in Yunnan, China. Am. J. Bot. 2010, 97, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, P.; Woeste, K.E.; Chiocchini, F.; Olimpieri, I.; Tortolano, V.; Clark, J.; Hemery, E.G.; Mapelli, S.; Malvolti, M.E. Long-term human impacts on genetic structure of Italian walnut inferred by SSR markers. Tree Genet. Genomes 2011, 7, 707–723. [Google Scholar] [CrossRef]
- Wang, H.; Pan, G.; Ma, Q.; Zhang, J.; Pei, D. The genetic diversity and introgression of Juglans regia and Juglans sigillata in Tibet as revealed by SSR markers. Tree Genet. Genomes 2015, 11, 1–11. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Site | Population ID | Type | Sample Size | Longitude (E) | Latitude (N) | Elevation (m) |
|---|---|---|---|---|---|---|
| Zunyi, Guizhou | YW | Wild | 8 | 106°47′35.22″ | 27°18′18.29″ | 684 |
| Nanchong, Sichuan | SC | Wild | 8 | 105°55′30.52″ | 30°52′33.19″ | 437 |
| Linzhi, Xizang | XZ | Wild | 8 | 94°21′42.94″ | 29°38′50.75″ | 2995 |
| Tianshui, Gansu | GS | Wild | 8 | 106°00′35.96″ | 34°20′55.10″ | 1579 |
| Akesu, Xinjiang | XJ | Wild | 8 | 82°57′43.26″ | 41°43′04.46″ | 1072 |
| Longshan, Hunan | LS | Wild | 8 | 109°30′16.83″ | 29°13′21.52″ | 479 |
| Nanchang, Jiangxi | MT | Cultivated | 8 | 115°27′21.8″ | 28°44′32.38″ | 1235 |
| Guizhou | GZ | Cultivated | 8 | 104°40′37.64″ | 26°30′32.88″ | 1084 |
| Baoshan, Yunnan | BS | Cultivated | 8 | 98°47′8.29″ | 25°17′31.08″ | 1800 |
| Yuncheng, Shanxi | YC | Cultivated | 8 | 110°59′40.34″ | 35°01′59.86″ | 370 |
| Cili, Hunan | CL | Cultivated | 8 | 110°55′28.5″ | 29°23′41.65″ | 98 |
| Locus | Repeats | Primer Sequence (5′–3′) | GenBank Accession | NA | Size Range (bp) | Tm (°C) | PIC | Ho | He | HW |
|---|---|---|---|---|---|---|---|---|---|---|
| JR0082 | (AAAC)5 | F: AATTGCCACCAACGAACACG | JZ844947 | 5 | 144–160 | 53 | 0.642 | 0.694 | 0.694 | ** |
| R: TCGTTCCCCAGAAACTCTCCCCCAA | ||||||||||
| JR0160 | (TC)10 | F: TCTCGGATTTGGGCTGTGAC | JZ844948 | 6 | 276–282 | 53 | 0.695 | 0.476 | 0.662 | NS |
| R: TCCGGGACCCTCGTCTAATT | ||||||||||
| JR1165 | (AGAT)6 | F: CACGTAGCGTCCGTAATCGA | JZ844949 | 5 | 482–502 | 55 | 0.529 | 0.358 | 0.615 | NS |
| R: CAGCACCTCCACTAACTGCA | ||||||||||
| JR1739 | (GAGCCG)8 | F: GGATGTGGAGACGGCAAAGA | JZ844950 | 7 | 270–302 | 53 | 0.560 | 0.924 | 0.632 | *** |
| R: CGTCCACCCAAACCAAGAGA | ||||||||||
| JR1817 | (AC)11 | F: CCTCAGAGCCAACCATCCTT | JZ844951 | 5 | 371–381 | 55 | 0.606 | 0.576 | 0.660 | NS |
| R: AGAACAGAACCAGCGTCACA | ||||||||||
| JR2018 | (TC)10 | F: TCTCAACCTTGGCCTGCATT | JZ844952 | 4 | 268–278 | 55 | 0.583 | 0.762 | 0.661 | NS |
| R: CGAAAAGCCAACCTTCGCAA | ||||||||||
| JR2465 | (TC)10 | F: GTTCCTCTTTCCCCAGCCTC | JZ844953 | 4 | 309–317 | 53 | 0.518 | 0.021 | 0.607 | *** |
| R: TCTGGCCACCATTGTAGCTG | ||||||||||
| JR2510 | (ATTAT)5 | F: GGGGATGTTGGGGGTTGATT | JZ844954 | 2 | 315–320 | 52 | 0.282 | 0.435 | 0.344 | ND |
| R: ACTTGTGGAGGGGAGGAAGA | ||||||||||
| JR2600 | (GA)10 | F: TTGGGGAATCTGCAGCAGAG | JZ844955 | 4 | 135–141 | 51 | 0.524 | 0.872 | 0.601 | *** |
| R: TATTACACATGCCGCAGCCA | ||||||||||
| JR2873 | (GGGGCG)5 | F: GGTTAGGGTAGCGGGTTCG | JZ844956 | 4 | 231–249 | 55 | 0.572 | 0.929 | 0.651 | * |
| R: AGCGACGATGGAAACGAACT | ||||||||||
| JR3147 | (CTAT)6 | F: CAGCACCTCCACTAACTGCA | JZ844957 | 3 | 480–488 | 55 | 0.479 | 0.513 | 0.575 | NS |
| R: CACGTAGCGTCCGTAATCGA | ||||||||||
| JR3434 | (GTAT)5 | F: CCGCCCAGCAGATTGTCATA | JZ844958 | 2 | 276–280 | 55 | 0.342 | 0.142 | 0.441 | *** |
| R:CGTCCCCTCAAGTTCTTGCT | ||||||||||
| JR3608 | (ATTA)5 | F: CCCCTCCCCCATTTCTTGAC | JZ844959 | 4 | 276–288 | 55 | 0.436 | 0.411 | 0.525 | NS |
| R: TCATGTAACATCATTCACCAACCA | ||||||||||
| JR3773 | (CTGT)5 | F: GGTGGTTTGACCCTTAATTCTGT | JZ844960 | 3 | 173–181 | 55 | 0.345 | 0.299 | 0.379 | ND |
| R: ACCCTGCCACAATGACCAAA | ||||||||||
| JR4051 | (TCTT)5 | F: TGAGGCTATAACCACCCCCT | JZ844961 | 4 | 206–218 | 55 | 0.483 | 0.559 | 0.543 | NS |
| R: GGCAACCAAGAGAAGCAAGG | ||||||||||
| JR4324 | (AT)10 | F: AGTGGCTTCTTGATTGTGCCT | JZ844962 | 4 | 266–274 | 55 | 0.248 | 0.260 | 0.275 | ND |
| R: GCTGTCCTCATCGTTTGTGC | ||||||||||
| JR4616 | (AGAC)5 | F: AGCCCTTTTGCATCGGCTAT | JZ844963 | 2 | 160–164 | 55 | 0.320 | 0.203 | 0.403 | ** |
| R: AGCTGACCGATCGATCAACA | ||||||||||
| JR4964 | (GGGA)5 | F: CTCGATCTGAACTCGGCTCC | JZ844965 | 4 | 214–226 | 52 | 0.461 | 0.314 | 0.515 | NS |
| R:TCTACTCTCTCCGCACCACA | ||||||||||
| JR4965 | (AC)10 | F: TGTGGCTTCGTTAGTGTTGTG | JZ844966 | 4 | 288–298 | 55 | 0.337 | 0.241 | 0.387 | ND |
| R: TCTTTTCCCTGAGTGGAGTTACA | ||||||||||
| JR49652 | (TG)10 | F: GCGCAGATCAATGAAAAGAGGG | JZ844967 | 3 | 266–270 | 55 | 0.268 | 0.234 | 0.306 | ND |
| R: TGTGGCTTCGTTAGTGTTGTG | ||||||||||
| JR5538 | (TG)10 | F: AGCTCACATCCAATCCAGCG | JZ844968 | 4 | 558–564 | 52 | 0.656 | 0.472 | 0.715 | NS |
| R:CCCCATCCCAAGAATCTCCC | ||||||||||
| JR5574 | (ATTT)5 | F:TGGTTAGTGACAGACCGCAG | JZ844986 | 4 | 200–296 | 55 | 0.530 | 0.522 | 0.609 | NS |
| R:CAGCAGCAGCAGTAGCAATG | ||||||||||
| JR6160 | (GA)10 | F: ACTTCAGGTTCCCAACGCAA | JZ844969 | 6 | 198–208 | 55 | 0.646 | 0.691 | 0.696 | NS |
| R: TAGAGGGAAGGTCTCCGGTG | ||||||||||
| JR6226 | (T)11g(A)10(AAT)5 | F: TGAGATGTTTGGCACGCTGA | JZ844970 | 3 | 238–244 | 55 | 0.402 | 0.881 | 0.515 | *** |
| R: AATGCCGTCGCCTACTTGAA | ||||||||||
| JR6439 | (TGCG)5 | F: TCGATGCGATCATCTCCGTG | JZ844971 | 3 | 148–156 | 52 | 0.393 | 0.224 | 0.515 | *** |
| R: CGGCACCAAAACAGAACTCG | ||||||||||
| JR6508 | (TCTT)5 | F: CGTCGATGACAAGTCCGGAT | JZ844972 | 4 | 267–279 | 55 | 0.443 | 0.417 | 0.517 | NS |
| R: CAGCTCTCAGACACACAGGG | ||||||||||
| JR6638 | (T)12cgtt(A)10 | F: CTGACAGACATGGAGGGTCG | JZ844973 | 2 | 222–224 | 55 | 0.280 | 0.016 | 0.339 | ND |
| R: ACAAACTATATTGTGCAAGAATCCAGT | ||||||||||
| JR6714 | (AT)6aa(AT)10 | F: TGGGGGCTCTTTCTTCCAAA | JZ844974 | 2 | 185–193 | 52 | 0.230 | 0.126 | 0.135 | ND |
| R: CCTTGCAAACATCATCCACACT | ||||||||||
| JR6742 | (TGTC)6 | F: AGCTCTAGCCTCTAGGGGTTC | JZ844975 | 3 | 247–255 | 55 | 0.584 | 0.394 | 0.663 | * |
| R: TCCCCAATTAATTGCAAACACCA | ||||||||||
| JR6926 | (CAAC)5 | F: GGAAAGGCATTGCAGAGCAC | JZ844976 | 2 | 176–180 | 55 | 0.341 | 0.116 | 0.438 | *** |
| R: GGCAGAGCAAGAGACTTCGT | ||||||||||
| JR7171 | ((TCCC)5 | F: ACCTAATCCACGTGCGACAG | JZ844977 | 4 | 327–339 | 53 | 0.373 | 0.712 | 0.473 | *** |
| R: GCTCTTCCTCCGTCCTCAAC | ||||||||||
| JR7363 | (AT)10 | F: GGCCATCGAAAATAGCAAACGA | JZ844978 | 4 | 162–168 | 55 | 0.526 | 0.132 | 0.592 | *** |
| R: AGTGGCTTCTTGATTGTGCCT | ||||||||||
| JR7495 | (GTTG)5 | F: GGCAGAGCAAGAGACTTCGT | JZ844979 | 2 | 248–254 | 55 | 0.375 | 0.785 | 0.503 | *** |
| R: GGAAAGGCATTGCAGAGCAC | ||||||||||
| JR74952 | (A)10c(AT)7 | F:ACGATCCCCTTTGCTTGCAT | JZ844980 | 2 | 174–176 | 55 | 0.168 | 0.122 | 0.138 | ND |
| R:AGGGCAGCCACATATGATCA | ||||||||||
| JR7544 | (ATACG)5 | F: CCTCGGGTCCACCTTTCTTC | JZ844981 | 4 | 192–207 | 55 | 0.455 | 0.187 | 0.506 | *** |
| R: TCGCTGCGAAACTCTTGAGT | ||||||||||
| JR8058 | (AG)10 | F: TTGTGTTGCTGGGTCTTCGT | JZ844982 | 3 | 172–184 | 55 | 0.150 | 0.053 | 0.052 | ND |
| R: AGAAAAGGTGCGCAGTGAGA | ||||||||||
| JR8815 | (AGTCT)5 | F: TTCTGGGATGAGGAGGAGGG | JZ844983 | 3 | 221–231 | 55 | 0.408 | 0.077 | 0.504 | *** |
| R: CCGAAATCACGCAGGAAAGC | ||||||||||
| JR9306 | (GA)11 | F: GGTGACCACAACACGCTACT | JZ844984 | 3 | 218–224 | 52 | 0.213 | 0.255 | 0.231 | ND |
| R: ACCTCTTGTGCCTCTGAACG | ||||||||||
| JR9632 | (CGAGCA)8 | F: CCGTCTCCGCCTTTTACCTT | JZ844985 | 5 | 254–272 | 52 | 0.471 | 0.370 | 0.519 | NS |
| R: AGCTCAACGGTCAAGGAAGG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, M.; Zhang, T.; Hu, Y.; Zhou, H.; Woeste, K.E.; Zhao, P. De Novo Assembly and Characterization of Bud, Leaf and Flowers Transcriptome from Juglans Regia L. for the Identification and Characterization of New EST-SSRs. Forests 2016, 7, 247. https://doi.org/10.3390/f7100247
Dang M, Zhang T, Hu Y, Zhou H, Woeste KE, Zhao P. De Novo Assembly and Characterization of Bud, Leaf and Flowers Transcriptome from Juglans Regia L. for the Identification and Characterization of New EST-SSRs. Forests. 2016; 7(10):247. https://doi.org/10.3390/f7100247
Chicago/Turabian StyleDang, Meng, Tian Zhang, Yiheng Hu, Huijuan Zhou, Keith E. Woeste, and Peng Zhao. 2016. "De Novo Assembly and Characterization of Bud, Leaf and Flowers Transcriptome from Juglans Regia L. for the Identification and Characterization of New EST-SSRs" Forests 7, no. 10: 247. https://doi.org/10.3390/f7100247