Dominant-Negative Proteins in Herpesviruses – From Assigning Gene Function to Intracellular Immunization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Scope
2 .The Way to Assign Herpesviral Gene Functions
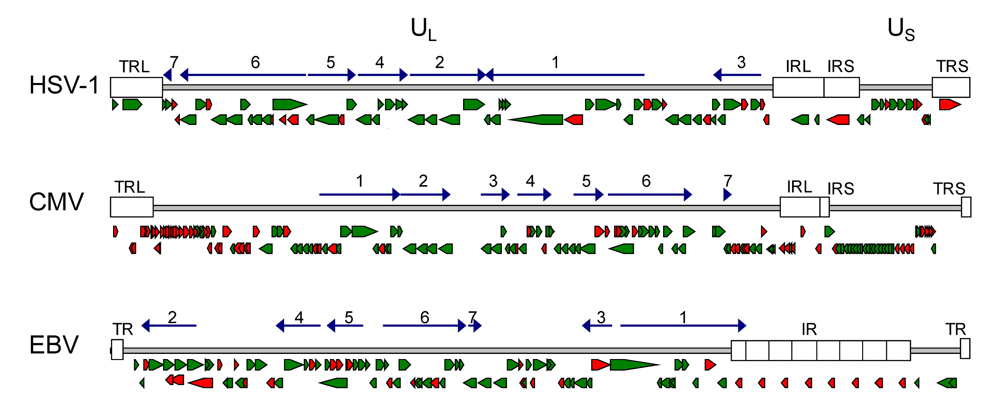
2.1. Conservation of genes and protein function in herpesviruses
2.2. Strategies to identify herpesviral gene function

2.2.2. Identification of gene function via homology screening
3. Dominant-Negative Proteins
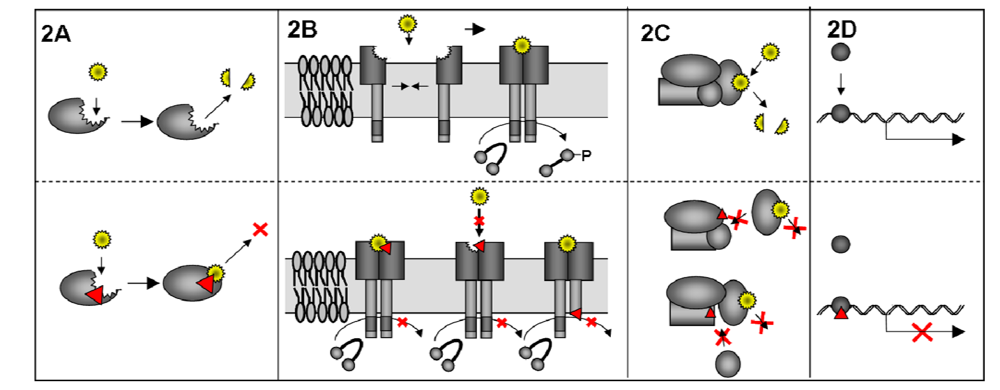
3.1. Mechanism of DN proteins


4. Elucidating Herpesvirus Biology with the Help of DN Proteins
4.1. Identification of pathways with cellular DN proteins
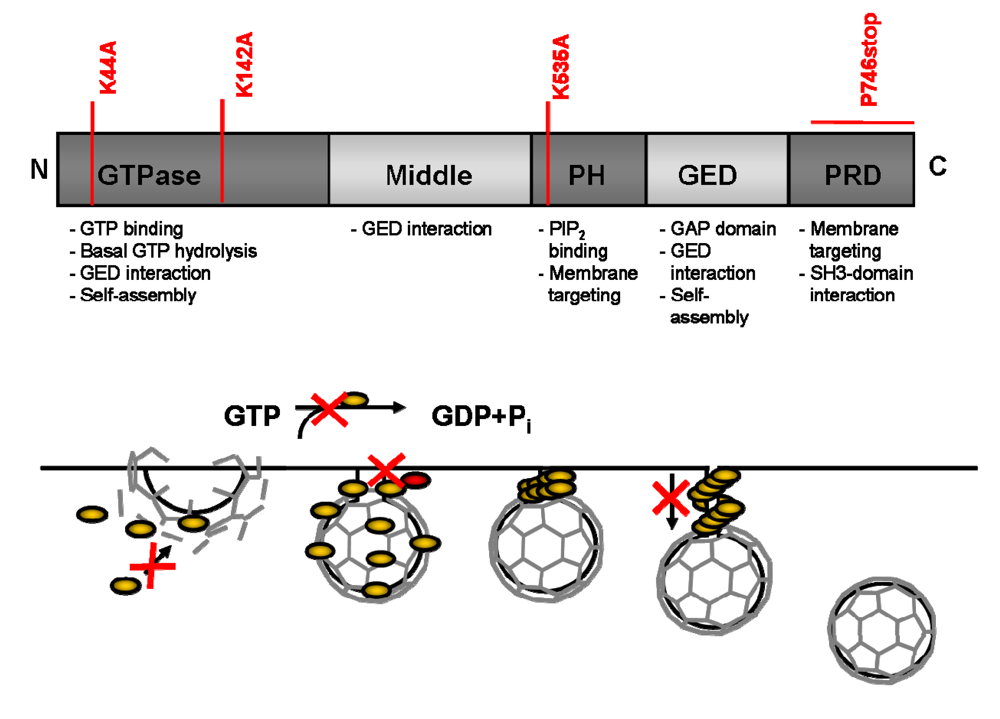
5. Design of DN Proteins
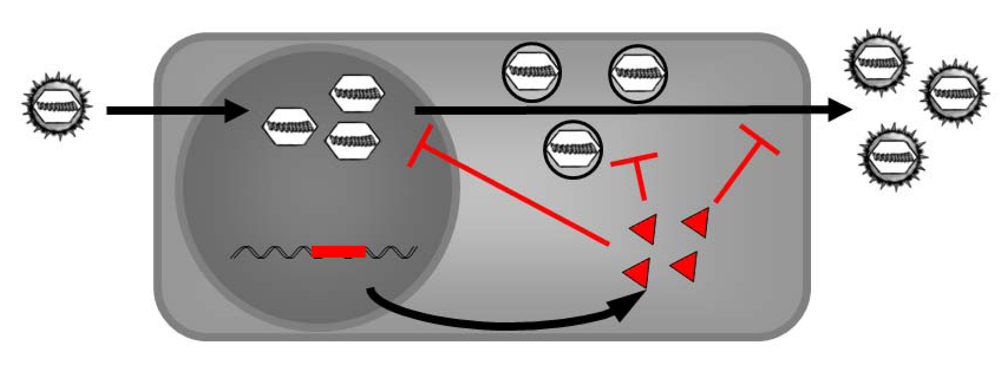
6. Intracellular Immunization - Using the Antiviral Properties of DN Proteins

7. Conclusions and Perspectives
Acknowledgments
References and Notes
- Roizman, B.; Baines, J. The Diversity and Unity of Herpes-Viridae. Comp. Immunol. Microb. 1991, 2, 63–79. [Google Scholar] [CrossRef]
- Fu, M.; Deng, R.; Wang, J.; Wang, X. Whole-Genome Phylogenetic Analysis of Herpesviruses. Acta Virol. 2008, 1, 31–40. [Google Scholar]
- Davison, A.J. Evolution of the herpesviruses. Vet. Microbiol. 2002, 1-2, 69–88. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Rixon, F. J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 1, 90–104. [Google Scholar] [CrossRef]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Immunol. Today 2000, 9, 447–455. [Google Scholar] [CrossRef]
- Mori, I.; Nishiyama, Y. Herpes simplex virus and varicella-zoster virus: why do these human alphaherpesviruses behave so differently from one another? Rev. Med. Virol. 2005, 6, 393–406. [Google Scholar] [CrossRef]
- Andoniou, C.E.; Degli-Esposti, M.A. Insights into the mechanisms of CMV-mediated interference with cellular apoptosis. Immunol. Cell. Biol. 2006, 1, 99–106. [Google Scholar] [CrossRef]
- Matis, J.; Kudelova, M. Early shutoff of host protein synthesis in cells infected with herpes simplex viruses. Acta. Virol. 2001, 5-6, 269–277. [Google Scholar]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F.Y. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U S A 2003, 24, 14223–14228. [Google Scholar] [CrossRef]
- Herskowitz, I. Functional Inactivation of Genes by Dominant Negative Mutations. Nature 1987, 6136, 219–222. [Google Scholar] [CrossRef]
- Wang, N.; Baldi, P.F.; Gaut, B.S. Phylogenetic analysis, genome evolution and the rate of gene gain in the Herpesviridae. Mol. Phylogenet. Evol. 2007, 3, 1066–1075. [Google Scholar] [CrossRef]
- Montague, M.G.; Hutchison, C.A. Gene content phylogeny of herpesviruses. Proc. Natl. Acad. Sci. U S A 2000, 10, 5334–5339. [Google Scholar] [CrossRef]
- Pellett, P.E.; Roizman, B. The Family Herpesviridae: A Brief Introduction. 2007, 66, 2479–2499. [Google Scholar]
- Salsman, J.; Zimmerman, N.; Chen, T.; Domagala, M.; Frappier, L. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. Plos Pathogens 2008, 7. [Google Scholar]
- Sweet, C.; Ball, K.; Morley, P.J.; Guilfoyle, K.; Kirby, M. Mutations in the temperature-sensitive murine cytomegalovirus (MCMV) mutants tsm5 and tsm30: A study of genes involved in immune evasion, DNA packaging and processing, and DNA replication. J. Med. Virol. 2007, 3, 285–299. [Google Scholar] [CrossRef]
- Benyesh-Melnick, M.; Schaffer, P.A.; Courtney, R.J.; Esparza, J.; Kimura, S. Viral gene functions expressed and detected by temperature-sensitive mutants of herpes simplex virus. Cold Spring Harb.Symp. Quant. Biol. 1975, 731–746. [Google Scholar]
- Akel, H.M.O.; Sweet, C. Isolation and Preliminary Characterization of 25 Temperature-Sensitive Mutants of Mouse Cytomegalovirus. FEMS Microbiol. Lett. 1993, 3, 253–260. [Google Scholar] [CrossRef]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; Baldanti, F.; Gerna, G. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 18, 10023–10033. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Post, L.E.; Roizman, B. Molecular Engineering of the Herpes-Simplex Virus Genome - Insertion of A Second L-S Junction Into the Genome Causes Additional Genome Inversions. Cell 1980, 1, 243–255. [Google Scholar] [CrossRef]
- Messerle, M.; Crnkovic, I.; Hammerschmidt, W.; Ziegler, H.; Koszinowski, U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. U S A 1997, 26, 14759–14763. [Google Scholar] [CrossRef]
- Brune, W.; Messerle, M.; Koszinowski, U.H. Forward with BACs - new tools for herpesvirus genomics. Trends Genet. 2000, 6, 254–259. [Google Scholar] [CrossRef]
- Adler, H.; Messerle, M.; Koszinowski, U.H. Cloning of herpesviral genomes as bacterial artificial chromosomes. Rev. Med. Virol. 2003, 2, 111–121. [Google Scholar] [CrossRef]
- Brune, W.; Menard, C.; Hobom, U.; Odenbreit, S.; Messerle, M.; Koszinowski, U.H. Rapid identification of essential and nonessential herpesvirus genes by direct transposon mutagenesis. Nature Biotech. 1999, 4, 360–364. [Google Scholar]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U S A 2003, 21, 12396–12401. [Google Scholar] [CrossRef]
- Hobom, U.; Brune, W.; Messerle, M.; Hahn, G.; Koszinowski, U.H. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: Mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 2000, 17, 7720–7729. [Google Scholar] [CrossRef]
- Smith, G.A.; Enquist, L.W. Construction and transposon mutagenesis in Escherichica coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J. Virol. 1999, 8, 6405–6414. [Google Scholar]
- Menard, C.; Wagner, M.; Ruzsics, Z.; Holak, K.; Brune, W.; Campbell, A.E.; Koszinowski, U.H. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 2003, 10, 5557–5570. [Google Scholar] [CrossRef]
- Brune, W.; Menard, C.; Heesemann, J.; Koszinowski, U.H. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 2001, 5502, 303–305. [Google Scholar] [CrossRef]
- Ruzsics, Z.; Wagner, M.; Osterlehner, A.; Cook, J.; Koszinowski, U.; Burgert, H.G. Transposon-assisted cloning and traceless mutagenesis of adenoviruses: Development of a novel vector based on species D. J. Virol. 2006, 16, 8100–8113. [Google Scholar] [CrossRef]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 2, 191–197. [Google Scholar]
- Bubic, I.; Wagner, M.; Krmpotic, A.; Saulig, T.; Kim, S.; Yokoyama, W.M.; Jonjic, S.; Koszinowski, U.H. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J. Virol. 2004, 14, 7536–7544. [Google Scholar] [CrossRef]
- Champier, G.; Hantz, S.; Couvreux, A.; Stuppfler, S.; Mazeron, M.C.; Bouaziz, S.; Denis, F.; Alain, S. New functional domains of human cytomegalovirus pUL89 predicted by sequence analysis and three-dimensional modelling of the catalytic site DEXDc. Antiviral Ther. 2007, 2, 217–232. [Google Scholar]
- Holzerlandt, R.; Orengo, C.; Kellam, P.; Alba, M.M. Identification of new herpesvirus gene homologs in the human genome. Genome Res. 2002, 11, 1739–1748. [Google Scholar] [CrossRef]
- Bottcher, S.; Klupp, B.G.; Granzow, H.; Fuchs, W.; Michael, K.; Mettenleiter, T.C. Identification of a 709-amino-acid internal nonessential region within the essential conserved tegument protein (p)UL36 of pseudorabies virus. J. Virol. 2006, 19, 9910–9915. [Google Scholar] [CrossRef]
- Veitia, R.A. Exploring the molecular etiology of dominant-negative mutations. Plant Cell 2007, 12, 3843–3851. [Google Scholar] [CrossRef]
- Veitia, R.A. Dominant negative factors in health and disease. J. Pathol. 2009, 218, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Yamanaka, M.; Imamura, K.; Tanaka, Y.; Nara, A.; Yoshimori, T.; Yokota, S.; Himeno, M. A dominant negative form of the AAA ATPase SKD1/VPS4 impairs membrane trafficking out of endosomal/lysosomal compartments: class E vps phenotype in mammalian cells. J. Cell. Sci. 2003, 2, 401–414. [Google Scholar] [CrossRef]
- Hendershot, L.M.; Wei, J.Y.; Gaut, J.R.; Melnick, J.; Aviel, S.; Argon, Y. Inhibition of Immunoglobulin Folding and Secretion by Dominant-Negative Bip Atpase Mutants. Mol. Biol. Cell. 1995, 375–375. [Google Scholar]
- Tabancay, A.P.; Gau, C.L.; Machado, I.M.P.; Uhlmann, E.J.; Gutmann, D.H.; Guo, L.; Tamanoi, F. Identification of dominant negative mutants of Rheb GTPase and their use to implicate the involvement of human Rheb in the activation of p70S6K. J. Biol. Chem. 2003, 41, 39921–39930. [Google Scholar] [CrossRef]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 241–255. [Google Scholar] [CrossRef]
- Rodriguez-Frade, J.M.; Vila-Coro, A.J.; de Ana, A.M.; Albar, J.P.; Martinez, A.; Mellado, M. The chemokine monocyte chemoattractant protein-1 induces functional responses through dimerization of its receptor CCR2. Proc. Natl. Acad. Sci. U S A 1999, 7, 3628–3633. [Google Scholar] [CrossRef]
- Caldenhoven, E.; vanDijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.M.; Lammers, J.W.J.; Koenderman, L.; deGroot, R.P. STAT3 beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 1996, 22, 13221–13227. [Google Scholar]
- Barren, B.; Artemyev, N.O. Mechanisms of dominant negative G-protein alpha subunits. J. Neurosci. Res. 2007, 16, 3505–3514. [Google Scholar] [CrossRef]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X.B. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 13, 2330–2338. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.Q.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 6669, 806–811. [Google Scholar] [CrossRef]
- Hinshaw, J.E. Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Biol. 2000, 16, 483–519. [Google Scholar] [CrossRef] [PubMed]
- Damke, H.; Baba, T.; Warnock, D.E.; Schmid, S.L. Induction of Mutant Dynamin Specifically Blocks Endocytic Coated Vesicle Formation. J. Cell. Biol. 1994, 4, 915–934. [Google Scholar] [CrossRef]
- Damke, H.; Binns, D.D.; Ueda, H.; Schmid, S.L.; Baba, T. Dynamin GTPase domain mutants block endocytic vesicle formation at morphologically distinct stages. Mol. Biol. Cell. 2001, 9, 2578–2589. [Google Scholar]
- Lee, A.; Frank, D.W.; Marks, M.S.; Lemmon, M.A. Dominant-negative inhibition of receptor-mediated endocytosis by a dynamin-1 mutant with a defective pleckstrin homology domain. Curr. Biol. 1999, 5, 261–264. [Google Scholar] [CrossRef]
- Achiriloaie, M.; Barylko, B.; Albanesi, J.P. Essential role of the dynamin pleckstrin homology domain in receptor-mediated endocytosis. Mol. Cell. Biol. 1999, 2, 1410–1415. [Google Scholar]
- Szaszak, M.; Gaborik, Z.; Turu, G.; McPherson, P.S.; Clark, A.J.L.; Catt, K.J.; Hunyady, L. Role of the proline-rich domain of dynamin-2 and its interactions with Src homology 3 domains during endocytosis of the AT(1) angiotensin receptor. J. Biol. Chem. 2002, 24, 21650–21656. [Google Scholar] [CrossRef]
- Marks, B.; Stowell, M.H.B.; Vallis, Y.; Mills, I.G.; Gibson, A.; Hopkins, C.R.; McMahon, H.T. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 2001, 6825, 231–235. [Google Scholar] [CrossRef]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Krausslich, H.G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 3, 1581–1594. [Google Scholar] [CrossRef]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and caveolin-independent entry of human papillomavirus type 16--involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 2008, 10, e3313. [Google Scholar] [CrossRef]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 2008, 11, 1653–1668. [Google Scholar] [CrossRef]
- Cheshenko, N.; Liu, W.; Satlin, L.M.; Herold, B.C. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J. Biol. Chem. 2005, 35, 31116–31125. [Google Scholar] [CrossRef]
- Veettil, M.V.; Sharma-Walia, N.; Sadagopan, S.; Raghu, H.; Sivakumar, R.; Naranatt, P.P.; Chandran, B. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 2006, 23, 11432–11446. [Google Scholar] [CrossRef]
- Sharma-Walia, N.; Naranatt, P.P.; Krishnan, H.H.; Zeng, L.; Chandran, B. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-Rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 2004, 8, 4207–4223. [Google Scholar] [CrossRef]
- Krishnan, H.H.; Sharma-Walia, N.; Streblow, D.N.; Naranatt, P.P.; Chandran, B. Focal adhesion kinase is critical for entry of Kaposi's sarcoma-associated herpesvirus into target cells. J. Virol. 2006, 3, 1167–1180. [Google Scholar] [CrossRef]
- Advani, S.J.; Weichselbaum, R.R.; Roizman, B. The role of cdc2 in the expression of herpes simplex virus genes. Proc. Natl. Acad. Sci. U S A 2000, 20, 10996–11001. [Google Scholar] [CrossRef]
- Gu, H.D.; Roizman, B. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. U S A 2003, 15, 8963–8968. [Google Scholar] [CrossRef]
- Cicin-Sain, L.; Ruzsics, Z.; Podlech, J.; Bubic, I.; Menard, C.; Jonjic, S.; Reddehase, M.J.; Koszinowski, U.H. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J. Virol. 2008, 5, 2056–2064. [Google Scholar] [CrossRef]
- Walters, M.S.; Hall, K.T.; Whitehouse, A. The herpesvirus saimiri open reading frame (ORF) 50 (Rta) protein contains an AT hook required for binding to the ORF 50 response element in delayed-early promoters. J. Virol. 2004, 9, 4936–4942. [Google Scholar] [CrossRef]
- Rossetto, C.; Yamboliev, I.; Pari, G.S. Kaposi's Sarcoma-Associated Herpesvirus/Human Herpesvirus 8 K-bZIP modulates LANA mediated suppression of lytic origin-dependent DNA synthesis. J. Virol. 2009, 83, 8492–8501. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.E.; Taylor, T.J.; Knipe, D.M. A dominant-negative herpesvirus protein inhibits intranuclear targeting of viral proteins: Effects on DNA replication and late gene expression. J. Virol. 2000, 21, 10122–10131. [Google Scholar] [CrossRef]
- Watanabe, S.; Ono, E.; Shimizu, Y.; Kida, H. Mapping of transregulatory domains of pseudorabies virus early protein 0 and identification of its dominant-negative mutant. Arch. Virol. 1996, 6, 1001–1009. [Google Scholar] [CrossRef]
- Kim, S.K.; Ahn, B.C.; Albrecht, R.A.; O'Callaghan, D.J. The unique IR2 protein of equine herpesvirus 1 negatively regulates viral gene expression. J. Virol. 2006, 10, 5041–5049. [Google Scholar] [CrossRef]
- Kirchmaier, A.L.; Sugden, B. Dominant-negative inhibitors of EBNA-1 of Epstein-Barr virus. J. Virol. 1997, 3, 1766–1775. [Google Scholar]
- Marintcheva, B.; Weller, S.K. Existence of transdominant and potentiating mutants of UL9, the herpes simplex virus type 1 origin-binding protein, suggests that levels of UL9 protein may be regulated during infection. J. Virol. 2003, 17, 9639–9651. [Google Scholar] [CrossRef]
- Olivo, P.D.; Nelson, N.J.; Challberg, M.D. Herpes-Simplex Virus-Dna Replication - the Ul9 Gene Encodes An Origin-Binding Protein. Proc. Natl. Acad. Sci. U S A 1988, 15, 5414–5418. [Google Scholar] [CrossRef]
- Yao, F.; Eriksson, E. A novel anti-herpes simplex virus type 1-specific herpes simplex virus type 1 recombinant. Hum. Gene Ther. 1999, 11, 1811–1818. [Google Scholar]
- Yao, F.; Eriksson, E. Inhibition of herpes simplex virus type 2 (HSV-2) viral replication by the dominant negative mutant polypeptide of HSV-1 origin binding protein. Antiviral Res. 2002, 2, 127–133. [Google Scholar] [CrossRef]
- Augustinova, H.; Hoeller, D.; Yao, F. The dominant-negative herpes simplex virus type 1 (HSV-1) recombinant CJ83193 can serve as an effective vaccine against wild-type HSV-1 infection in mice. J. Virol. 2004, 11, 5756–5765. [Google Scholar] [CrossRef]
- Brans, R.; Eriksson, E.; Yao, F. Immunization with a Dominant-Negative Recombinant HSV Type 1 Protects against HSV-1 Skin Disease in Guinea Pigs. J. Invest. Dermatol. 2008, 12, 2825–2832. [Google Scholar] [CrossRef]
- Prockop, D.J.; Kivirikko, K.I. Collagens - Molecular-Biology, Diseases, and Potentials for Therapy. Annu. Rev. Biochem. 1995, 403–434. [Google Scholar]
- Friedman, A.D.; Triezenberg, S.J.; Mcknight, S.L. Expression of A Truncated Viral Trans-Activator Selectively Impedes Lytic Infection by Its Cognate Virus. Nature 1988, 6189, 452–454. [Google Scholar] [CrossRef]
- Weber, P.C.; Wigdahl, B. Identification of Dominant-Negative Mutants of the Herpes-Simplex Virus Type-1 Immediate-Early Protein-Icp0. J. Virol. 1992, 4, 2261–2267. [Google Scholar]
- Borst, E.M.; Mathys, S.; Wagner, M.; Muranyi, W.; Messerle, M. Genetic evidence of an essential role for cytomegalovirus small capsid protein in viral growth. J. Virol. 2001, 3, 1450–1458. [Google Scholar] [CrossRef]
- Holzmayer, T.A.; Pestov, D.G.; Roninson, I.B. Isolation of Dominant Negative Mutants and Inhibitory Antisense Rna Sequences by Expression Selection of Random Dna Fragments. Nucleic Acids Res. 1992, 4, 711–717. [Google Scholar] [CrossRef]
- Steel, G.J.; Harley, C.; Boyd, A.; Morgan, A. A screen for dominant negative mutants of SEC18 reveals a role for the AAA protein consensus sequence in ATP hydrolysis. Mol. Biol. Cell 2000, 4, 1345–1356. [Google Scholar]
- Crowder, S.; Kirkegaard, K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nature Genet. 2005, 7, 701–709. [Google Scholar] [CrossRef]
- Cunningham, B.C.; Wells, J.A. High-Resolution Epitope Mapping of Hgh-Receptor Interactions by Alanine-Scanning Mutagenesis. Science 1989, 4908, 1081–1085. [Google Scholar]
- Thompson, D.K.; Garbers, D.L. Dominant-Negative Mutations of the Guanylyl Cyclase-A Receptor - Extracellular Domain Deletion and Catalytic Domain Point Mutations. J. Biol. Chem. 1995, 1, 425–430. [Google Scholar]
- Steel, G.J.; Harley, C.; Boyd, A.; Morgan, A. A screen for dominant negative mutants of SEC18 reveals a role for the AAA protein consensus sequence in ATP hydrolysis. Mol. Bio. Cell 2000, 4, 1345–1356. [Google Scholar]
- Hayes, F. Transposon-based strategies for microbial functional genomics and proteomics. Annu. Rev. Genet. 2003, 3–29. [Google Scholar] [CrossRef]
- Ruzsics, Z.; Koszinowski, U.H. Mutagenesis of the cytomegalovirus genome. In Human Cytomegalovirus. Shenk, T., Stinki, M.F., Eds.; 2008; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany. [Google Scholar]
- Bubeck, A.; Wagner, M.; Ruzsics, Z.; Lotzerich, M.; Iglesias, M.; Singh, I.R.; Koszinowski, U.H. Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J. Virol. 2004, 15, 8026–8035. [Google Scholar] [CrossRef]
- Lotzerich, M.; Ruzsics, Z.; Koszinowski, U.H. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J. Virol. 2006, 1, 73–84. [Google Scholar] [CrossRef]
- Rupp, B.; Ruzsics, Z.; Sacher, T.; Koszinowski, U.H. Conditional cytomegalovirus replication in vitro and in vivo. J. Virol. 2005, 1, 486–494. [Google Scholar] [CrossRef]
- Rupp, B.; Ruzsics, Z.; Buser, C.; Adler, B.; Walther, P.; Koszinowski, U.H. Random screening for dominant-negative mutants of the cytomegalovirus nuclear egress protein M50. J. Virol. 2007, 11, 5508–5517. [Google Scholar] [CrossRef]
- Granato, M.; Feederle, R.; Farina, A.; Gonnella, R.; Santarelli, R.; Hub, B.; Faggioni, A.; Delecluse, H.J. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 2008, 8, 4042–4051. [Google Scholar] [CrossRef]
- Lapik, Y.R.; Fernandes, C.J.; Lau, L.F.; Pestov, D.G. Physical and functional interaction between pes1 and bop1 in mammalian ribosome biogenesis. Mol. Cell 2004, 1, 17–29. [Google Scholar] [CrossRef]
- Osawa, M.; Erickson, H.P. Probing the domain structure of FtsZ by random insertional mutagenesis. Mol. Bio. Cell 2004, 292A–293A. [Google Scholar]
- Weber, E.; Koebnik, R. Domain structure of HrpE, the Hrp pilus subunit of Xanthomonas campestris pv. vesicatoria. J. Bacteriol. 2005, 17, 6175–6186. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Zhou, J.; Jones, C. Identification of functional domains within the bICP0 protein encoded by bovine herpesvirus 1. J. Gen. Virol. 2005, 879–886. [Google Scholar] [CrossRef]
- Cockrell, S.K.; Sanchez, M.E.; Erazo, A.; Homa, F.L. Role of the UL25 Protein in Herpes Simplex Virus DNA Encapsidation. J. Virol. 2009, 1, 47–57. [Google Scholar] [CrossRef]
- Lin, E.; Spear, P.G. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. U S A 2007, 32, 13140–13145. [Google Scholar] [CrossRef]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, reviews1017. [Google Scholar] [CrossRef] [PubMed]
- Best, S.; Le Tissier, P.; Towers, G.; Stoye, J.P. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 1996, 6594, 826–829. [Google Scholar] [CrossRef]
- Arnaud, F.; Varela, M.; Spencer, T.E.; Palmarini, M. Coevolution of endogenous betaretroviruses of sheep and their host. Cell Mol. Life Sci. 2008, 21, 3422–3432. [Google Scholar] [CrossRef]
- Palmarini, M.; Mura, M.; Spencer, T.E. Endogenous betaretroviruses of sheep: teaching new lessons in retroviral interference and adaptation. J. Gen. Virol. 2004, 1–13. [Google Scholar] [CrossRef]
- Baltimore, D. Gene-Therapy - Intracellular Immunization. Nature 1988, 6189, 395–396. [Google Scholar] [CrossRef]
- Trono, D.; Feinberg, M.B.; Baltimore, D. Hiv-1 Gag Mutants Can Dominantly Interfere with the Replication of the Wild-Type Virus. Cell 1989, 1, 113–120. [Google Scholar] [CrossRef]
- Bahner, I.; Sumiyoshi, T.; Kagoda, M.; Swartout, R.; Peterson, D.; Pepper, K.; Dorey, F.; Reiser, J.; Kohn, D.B. Lentiviral vector transduction of a dominant-negative rev gene into human CD34(+) hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol. Ther. 2007, 1, 76–85. [Google Scholar] [CrossRef]
- Wunner, W.H.; Pallatroni, C.; Curtis, P.J. Selection of genetic inhibitors of rabies virus. Arch. Virol. 2004, 8, 1653–1662. [Google Scholar]
- Jones, C.; Chowdhury, S. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim. Health Res. Rev. 2007, 2, 187–205. [Google Scholar]
- Gimeno, I.M. Marek's disease vaccines: A solution for today but a worry for tomorrow? Vaccine 2008, C31–C41. [Google Scholar] [CrossRef]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 1, 171–177. [Google Scholar] [CrossRef]
- Smith, C.A.; Deluca, N.A. Transdominant Inhibition of Herpes-Simplex Virus Growth in Transgenic Mice. Virology 1992, 2, 581–588. [Google Scholar] [CrossRef]
- Ono, E.; Sakoda, Y.; Taharaguchi, S.; Watanabe, S.; Tonomura, N.; Kida, H.; Shimizu, Y. Inhibition of Pseudorabies Virus-Replication by A Chimeric Trans-Gene Product Repressing Transcription of the Immediate-Early Gene. Virology 1995, 1, 128–140. [Google Scholar] [CrossRef]
- Ono, E.; Tasaki, T.; Kobayashi, T.; Taharaguchi, S.; Nikami, H.; Miyoshi, I.; Kasai, N.; Arikawa, J.; Kida, H.; Shimizu, Y. Resistance to pseudorabies virus infection in transgenic mice expressing the chimeric transgene that represses the immediate-early gene transcription. Virology 1999, 1, 72–78. [Google Scholar] [CrossRef]
- Tasaki, T.; Taharaguchi, S.; Kobayashi, T.; Yoshino, S.; Ono, E. Inhibition of pseudorabies virus replication by a dominant-negative mutant of early protein 0 expressed in a tetracycline-regulated system. Vet. Microbiol. 2001, 3, 195–203. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Mühlbach, H.; Mohr, C.A.; Ruzsics, Z.; Koszinowski, U.H. Dominant-Negative Proteins in Herpesviruses – From Assigning Gene Function to Intracellular Immunization. Viruses 2009, 1, 420-440. https://doi.org/10.3390/v1030420
Mühlbach H, Mohr CA, Ruzsics Z, Koszinowski UH. Dominant-Negative Proteins in Herpesviruses – From Assigning Gene Function to Intracellular Immunization. Viruses. 2009; 1(3):420-440. https://doi.org/10.3390/v1030420
Chicago/Turabian StyleMühlbach, Hermine, Christian A. Mohr, Zsolt Ruzsics, and Ulrich H. Koszinowski. 2009. "Dominant-Negative Proteins in Herpesviruses – From Assigning Gene Function to Intracellular Immunization" Viruses 1, no. 3: 420-440. https://doi.org/10.3390/v1030420