Mutations in the Non-Structural Protein-Coding Sequence of Protoparvovirus H-1PV Enhance the Fitness of the Virus and Show Key Benefits Regarding the Transduction Efficiency of Derived Vectors

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Infectious Molecular Plasmids
2.2. H-1PV-Based Vectors
2.3. Construction of H-1PV Split Vectors
2.4. Cell Cultures
2.5. Transfection Assays
2.6. Dual Luciferase Assay
2.7. Virus Production
2.8. Titration of Virus Stocks
2.9. Analysis of Plaque Size
2.10. Immunoblotting
2.11. Southern Blot Analysis
2.12. Northern Blot Analysis
2.13. Analysis of Virus Binding
2.14. Virus Uptake
2.15. Nuclear and Cytoplasmic Fractionation
3. Results
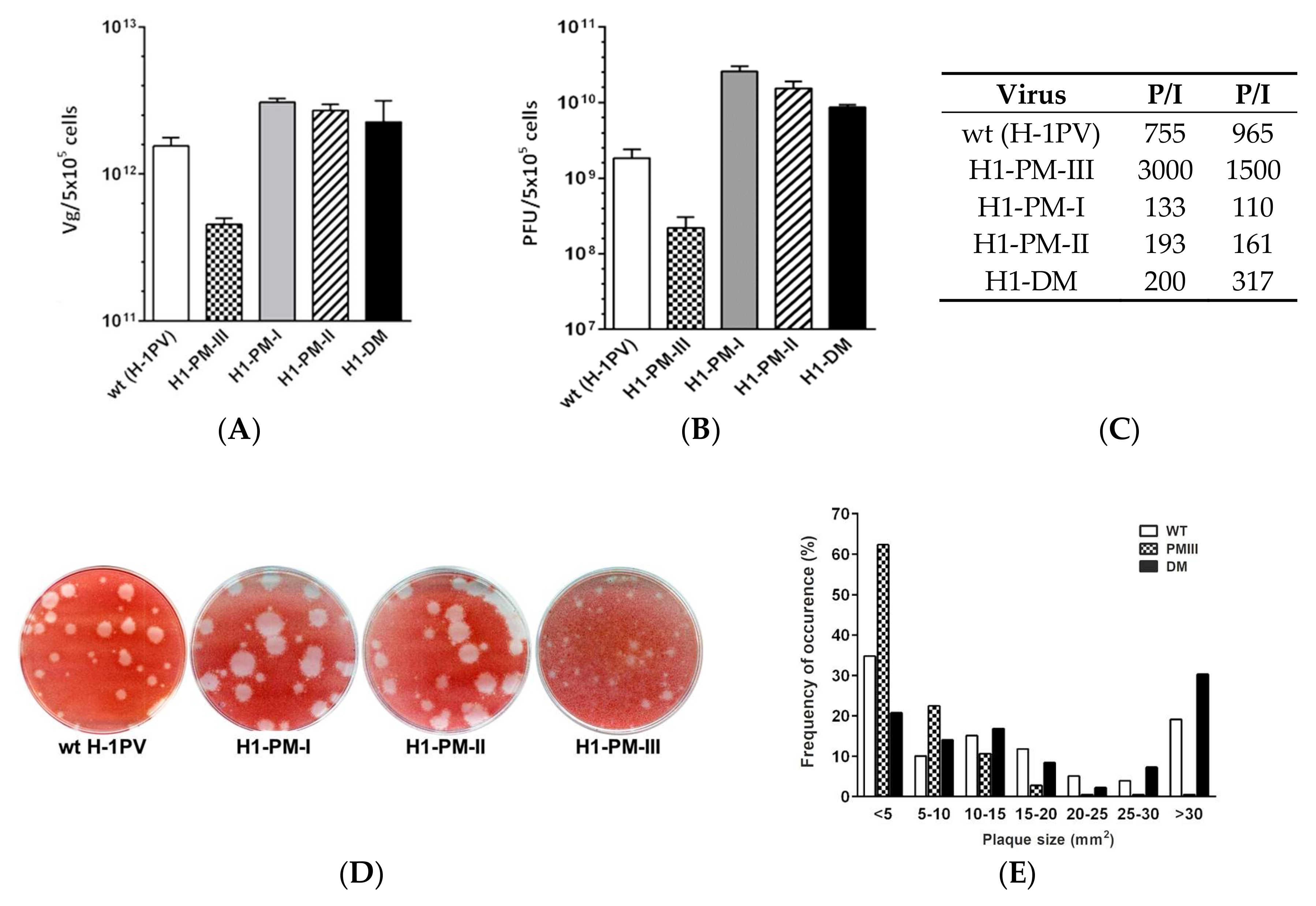
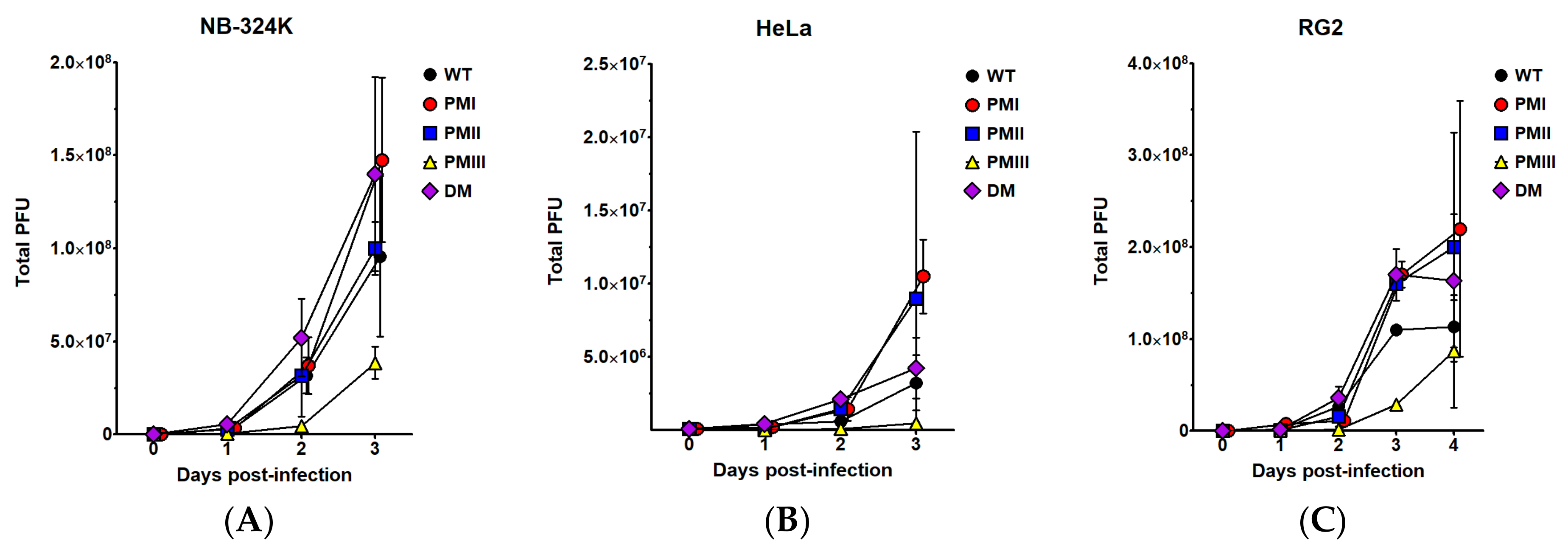
3.1. Production and Infectivity of H-1PV Mutants
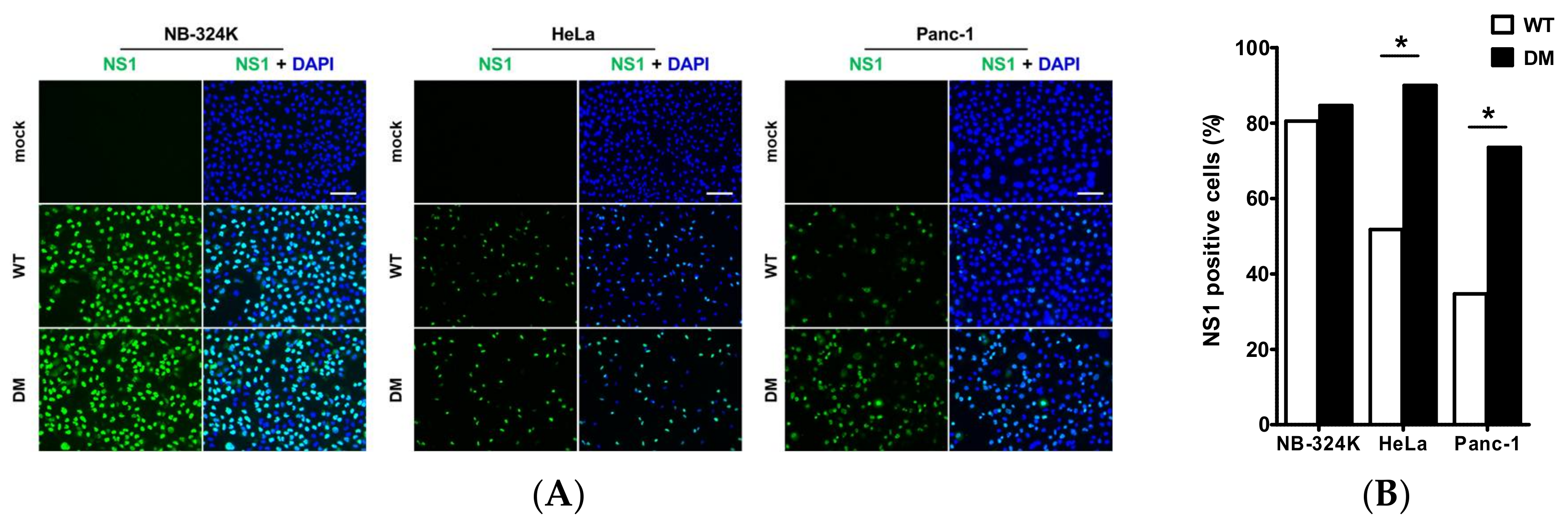
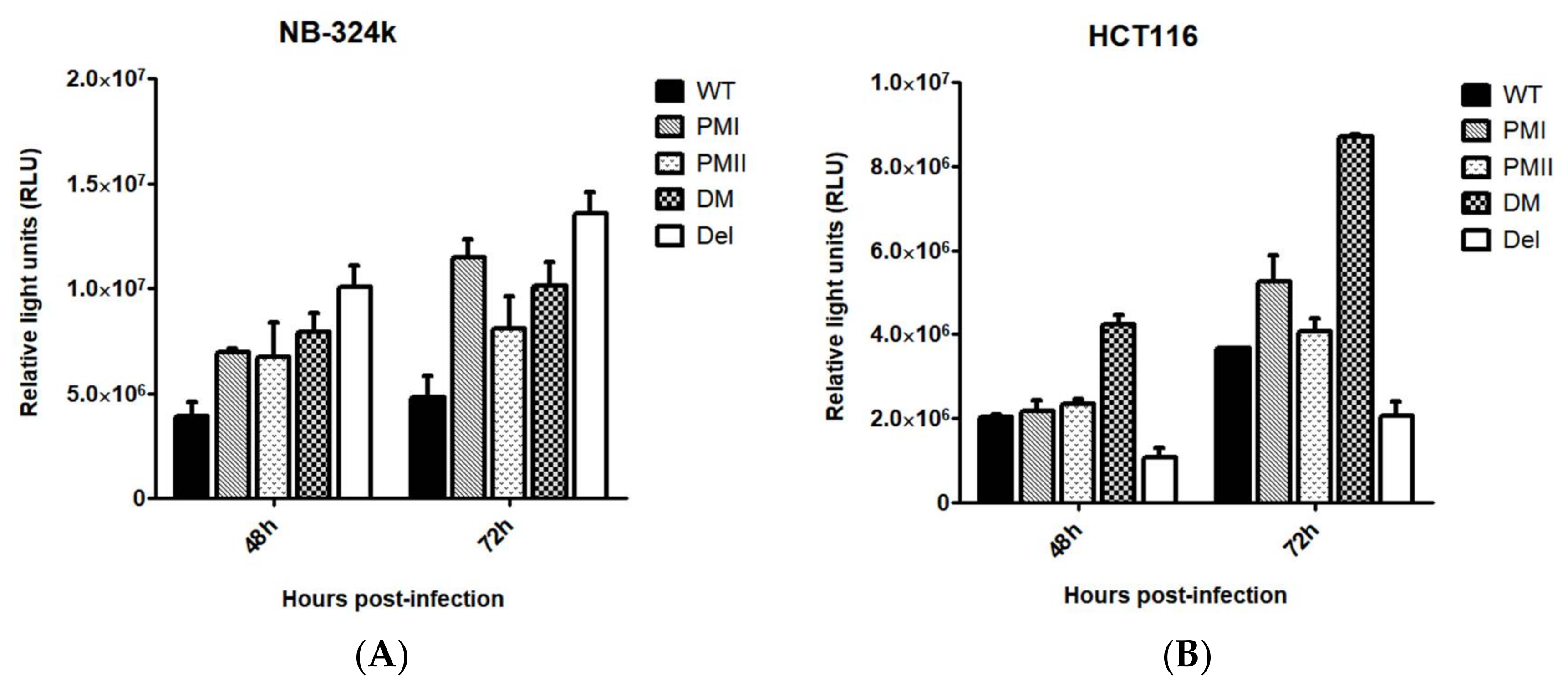
3.2. Enhanced Transduction Efficiency of H1-PM-I, H1-PM-II, and H1-DM-Based Vectors
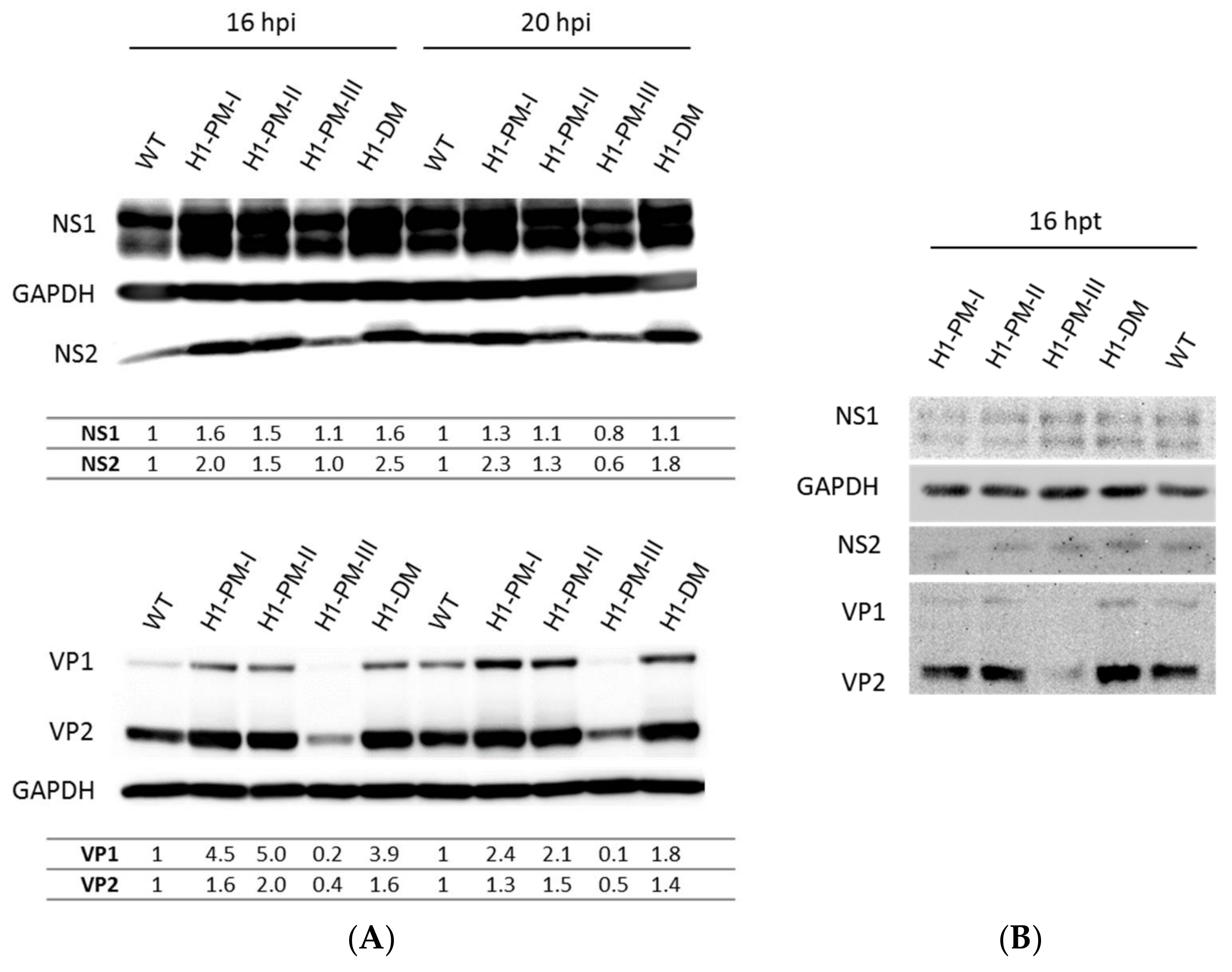
3.3. Enhanced Viral Protein Accumulation after Infection with the Fitness Mutants but Reduced Levels of Capsid Proteins with H1-PM-III
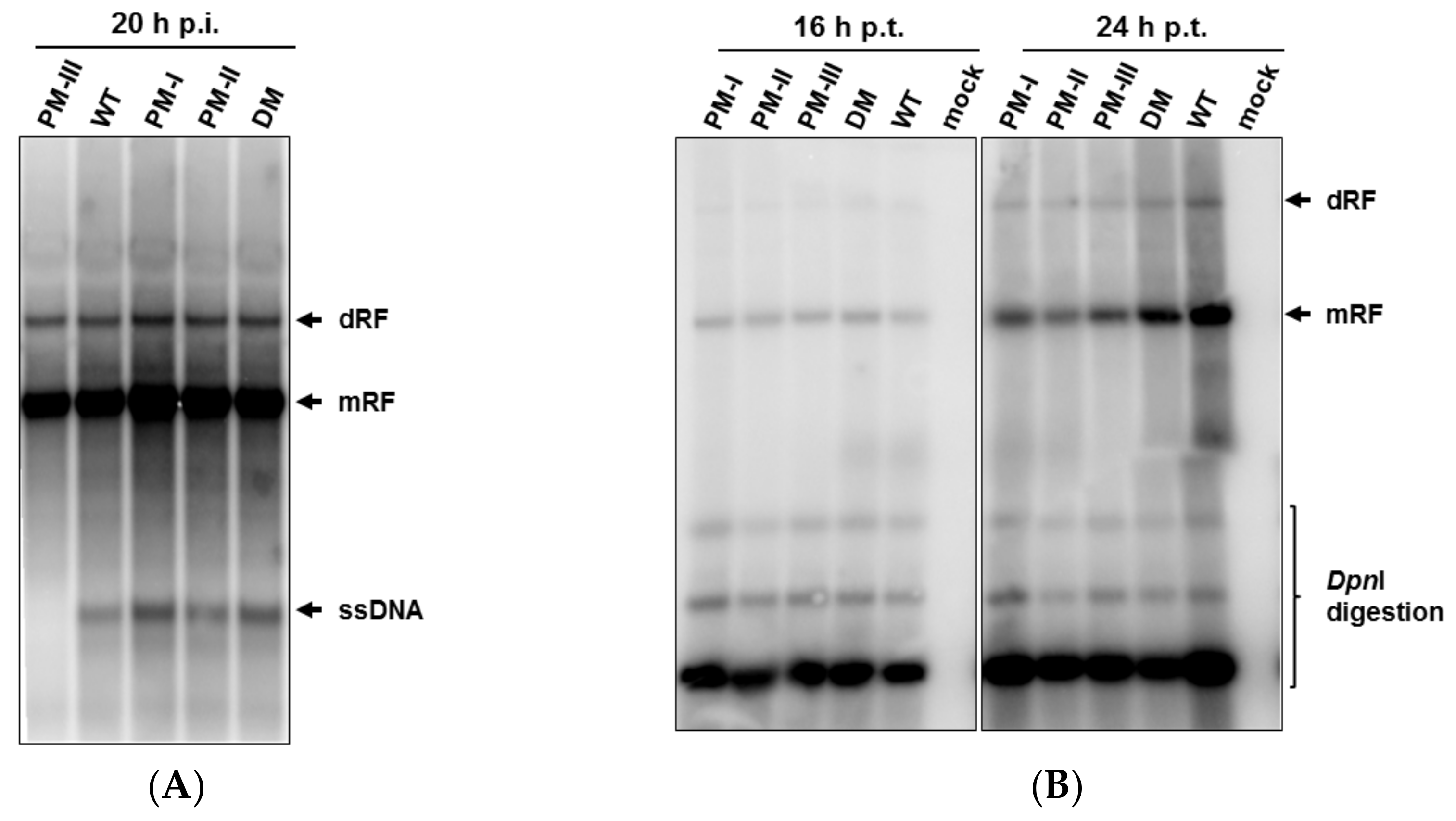
3.4. Enhanced Accumulation of Viral ssDNA after Infection with Fitness Mutants, but Severe Decrease of ssDNA Production in H1-PM-III-Infected Cells
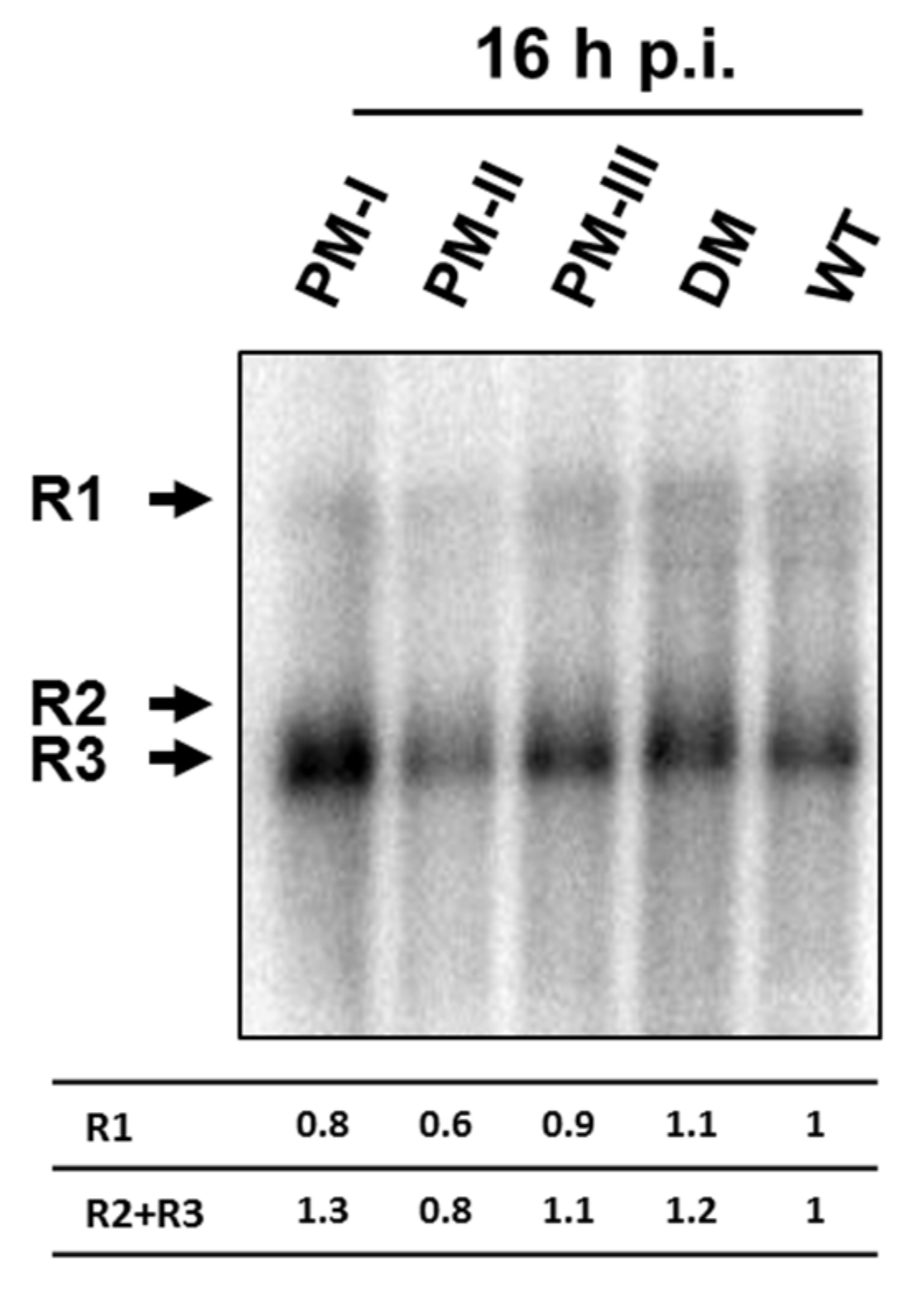
3.5. No Increased Accumulation of the Viral Transcripts after Infection with the Mutants Compared with Wild-Type H-1PV
3.6. Cis-Acting Effects of C2193A Mutation in H1-PM-III on Capsid Translation
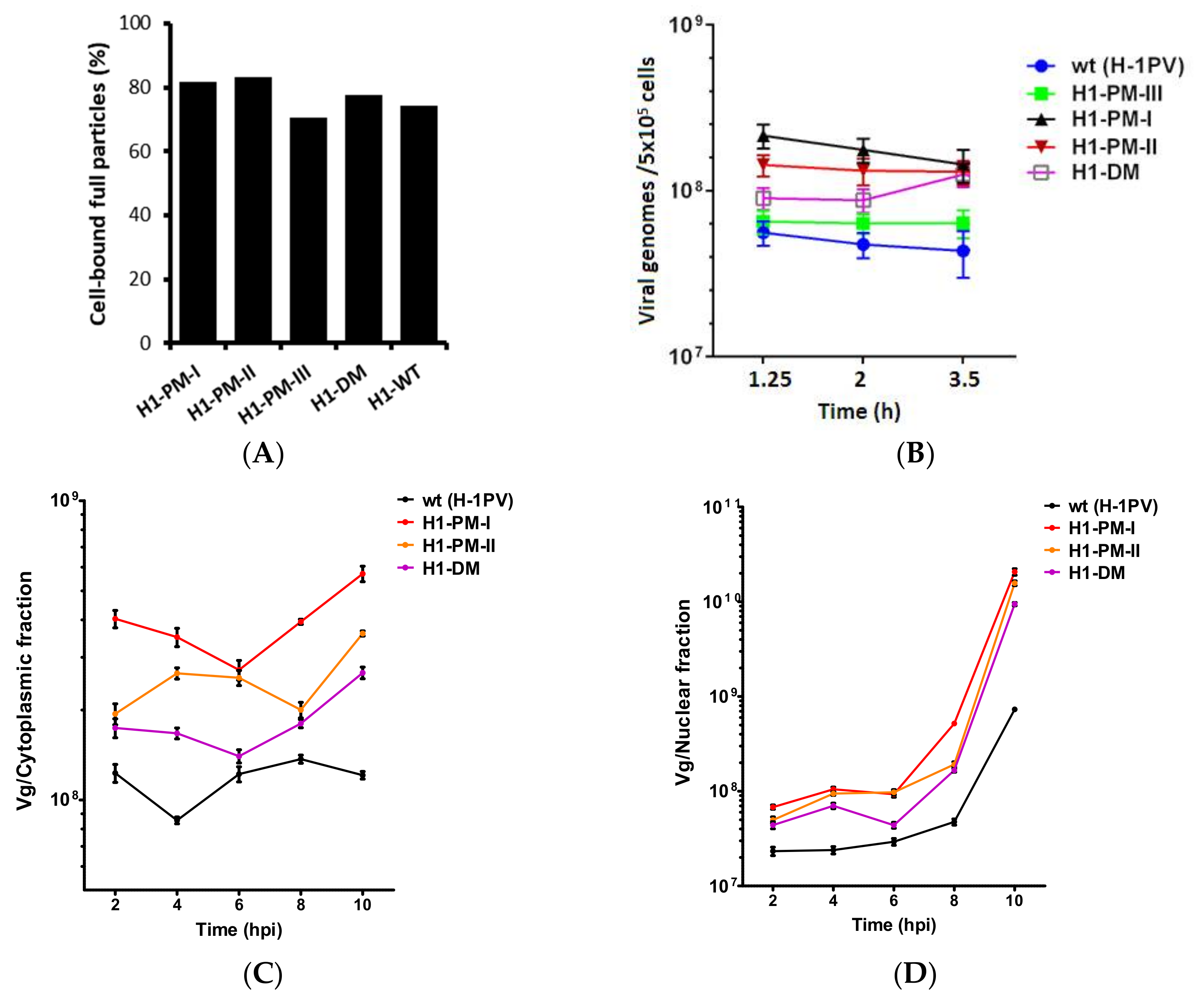
3.7. Improved Virus Binding and Cellular Uptake of the Fitness Mutants
4. Discussion
Key Benefits of PM-I, -II, and –DM for H-1PV Fitness and Transduction Efficiency of H-1PV-Based Vectors
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clemens, K.E.; Pintel, D.J. The two transcription units of the autonomous parvovirus minute virus of mice are transcribed in a temporal order. J. Virol. 1988, 62, 1448–1451. [Google Scholar] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 2007, 70, 183–232. [Google Scholar] [PubMed]
- Zadori, Z.; Szelei, J.; Tijssen, P. SAT: A late NS protein of porcine parvovirus. J. Virol. 2005, 79, 13129–13138. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rhode, S.L., III. Mutation of lysine 405 to serine in the parvovirus H-1 NS1 abolishes its functions for viral DNA replication, late promoter trans activation, and cytotoxicity. J. Virol. 1990, 64, 4654–4660. [Google Scholar] [PubMed]
- Caillet-Fauquet, P.; Perros, M.; Brandenburger, A.; Spegelaere, P.; Rommelaere, J. Programmed killing of human cells by means of an inducible clone of parvoviral genes encoding non-structural proteins. EMBO J. 1990, 9, 2989–2995. [Google Scholar] [PubMed]
- Nuesch, J.P.; Bar, S.; Rommelaere, J. Viral proteins killing tumor cells: New weapons in the fight against cancer. Cancer Biol. Ther. 2008, 7, 1374–1376. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Alternate splicing in a parvoviral nonstructural gene links a common amino-terminal sequence to downstream domains which confer radically different localization and turnover characteristics. Virology 1990, 177, 477–487. [Google Scholar] [CrossRef]
- Choi, E.Y.; Newman, A.E.; Burger, L.; Pintel, D. Replication of minute virus of mice DNA is critically dependent on accumulated levels of NS2. J. Virol. 2005, 79, 12375–12381. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; D’Abramo, A.M., Jr.; Carbonell, L.F.; Bratton, J.; Tattersall, P. The NS2 polypeptide of parvovirus MVM is required for capsid assembly in murine cells. Virology 1997, 231, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Naeger, L.K.; Cater, J.; Pintel, D.J. The small nonstructural protein (NS2) of the parvovirus minute virus of mice is required for efficient DNA replication and infectious virus production in a cell-type-specific manner. J. Virol. 1990, 64, 6166–6175. [Google Scholar] [PubMed]
- Naeger, L.K.; Salome, N.; Pintel, D.J. NS2 is required for efficient translation of viral mRNA in minute virus of mice-infected murine cells. J. Virol. 1993, 67, 1034–1043. [Google Scholar] [PubMed]
- Li, X.; Rhode, S.L., III. The parvovirus H-1 NS2 protein affects viral gene expression through sequences in the 3′ untranslated region. Virology 1993, 194, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Brandenburger, A.; Legendre, D.; Avalosse, B.; Rommelaere, J. NS-1 and NS-2 proteins may act synergistically in the cytopathogenicity of parvovirus MVMp. Virology 1990, 174, 576–584. [Google Scholar] [CrossRef]
- Brockhaus, K.; Plaza, S.; Pintel, D.J.; Rommelaere, J.; Salome, N. Nonstructural proteins NS2 of minute virus of mice associate in vivo with 14-3-3 protein family members. J. Virol. 1996, 70, 7527–7534. [Google Scholar] [PubMed]
- Young, P.J.; Jensen, K.T.; Burger, L.R.; Pintel, D.J.; Lorson, C.L. Minute virus of mice small nonstructural protein NS2 interacts and colocalizes with the Smn protein. J. Virol. 2002, 76, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Young, P.J.; Newman, A.; Jensen, K.T.; Burger, L.R.; Pintel, D.J.; Lorson, C.L. Minute virus of mice small non-structural protein NS2 localizes within, but is not required for the formation of, Smn-associated autonomous parvovirus-associated replication bodies. J. Gen. Virol. 2005, 86 Pt 4, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Bodendorf, U.; Cziepluch, C.; Jauniaux, J.C.; Rommelaere, J.; Salome, N. Nuclear export factor CRM1 interacts with nonstructural proteins NS2 from parvovirus minute virus of mice. J. Virol. 1999, 73, 7769–7779. [Google Scholar] [PubMed]
- Engelsma, D.; Valle, N.; Fish, A.; Salome, N.; Almendral, J.M.; Fornerod, M. A supraphysiological nuclear export signal is required for parvovirus nuclear export. Mol. Biol. Cell 2008, 19, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Eichwald, V.; Daeffler, L.; Klein, M.; Rommelaere, J.; Salome, N. The NS2 proteins of parvovirus minute virus of mice are required for efficient nuclear egress of progeny virions in mouse cells. J. Virol. 2002, 76, 10307–10319. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Pintel, D.J. Interaction between parvovirus NS2 protein and nuclear export factor Crm1 is important for viral egress from the nucleus of murine cells. J. Virol. 2002, 76, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Faisst, S.; Faisst, S.R.; Dupressoir, T.; Plaza, S.; Pujol, A.; Jauniaux, J.C.; Rhode, S.L.; Rommelaere, J. Isolation of a fully infectious variant of parvovirus H-1 supplanting the standard strain in human cells. J. Virol. 1995, 69, 4538–4543. [Google Scholar] [PubMed]
- Weiss, N.; Stroh-Dege, A.; Rommelaere, J.; Dinsart, C.; Salome, N. An in-frame deletion in the NS protein-coding sequence of parvovirus H-1PV efficiently stimulates export and infectivity of progeny virions. J. Virol. 2012, 86, 7554–7564. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Valle, N.; Gallego, J.M.; Perez, J.; Almendral, J.M. Enhanced cytoplasmic sequestration of the nuclear export receptor CRM1 by NS2 mutations developed in the host regulates parvovirus fitness. J. Virol. 2004, 78, 10674–10684. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schoning, T.; Husing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Dempe, S.; Lavie, M.; Struyf, S.; Bhat, R.; Verbeke, H.; Paschek, S.; Berghmans, N.; Geibig, R.; Rommelaere, J.; Van Damme, J.; et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: Implication of leucocyte recruitment. Cancer Immunol. Immunother. 2012, 61, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, P.; Bratton, J. Reciprocal productive and restrictive virus-cell interactions of immunosuppressive and prototype strains of minute virus of mice. J. Virol. 1983, 46, 944–955. [Google Scholar] [PubMed]
- Haag, A.; Menten, P.; Van Damme, J.; Dinsart, C.; Rommelaere, J.; Cornelis, J.J. Highly efficient transduction and expression of cytokine genes in human tumor cells by means of autonomous parvovirus vectors; generation of antitumor responses in recipient mice. Hum. Gene Ther. 2000, 11, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Zolotukhin, S.; Byrne, B.J.; Mason, E.; Zolotukhin, I.; Potter, M.; Chesnut, K.; Summerford, C.; Samulski, R.J.; Muzyczka, N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Kestler, J.; Neeb, B.; Struyf, S.; Van Damme, J.; Cotmore, S.F.; D’Abramo, A.; Tattersall, P.; Rommelaere, J.; Dinsart, C.; Cornelis, J.J. cis requirements for the efficient production of recombinant DNA vectors based on autonomous parvoviruses. Hum. Gene Ther. 1999, 10, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Wrzesinski, C.; Tesfay, L.; Salome, N.; Jauniaux, J.C.; Rommelaere, J.; Cornelis, J.; Dinsart, C. Chimeric and pseudotyped parvoviruses minimize the contamination of recombinant stocks with replication-competent viruses and identify a DNA sequence that restricts parvovirus H-1 in mouse cells. J. Virol. 2003, 77, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rhode, S.L., III. Nonstructural protein NS2 of parvovirus H-1 is required for efficient viral protein synthesis and virus production in rat cells in vivo and in vitro. Virology 1991, 184, 117–130. [Google Scholar] [CrossRef]
- Bar, S.; Rommelaere, J.; Nuesch, J.P. Vesicular transport of progeny parvovirus particles through ER and Golgi regulates maturation and cytolysis. PLoS Pathog. 2013, 9, e1003605. [Google Scholar] [CrossRef] [PubMed]
- Maroto, B.; Valle, N.; Saffrich, R.; Almendral, J.M. Nuclear export of the nonenveloped parvovirus virion is directed by an unordered protein signal exposed on the capsid surface. J. Virol. 2004, 78, 10685–10694. [Google Scholar] [CrossRef] [PubMed]
- Wolfisberg, R.; Kempf, C.; Ros, C. Late Maturation Steps Preceding Selective Nuclear Export and Egress of Progeny Parvovirus. J. Virol. 2016, 90, 5462–5474. [Google Scholar] [CrossRef] [PubMed]
- Daeffler, L.; Horlein, R.; Rommelaere, J.; Nuesch, J.P. Modulation of minute virus of mice cytotoxic activities through site-directed mutagenesis within the NS coding region. J. Virol. 2003, 77, 12466–12478. [Google Scholar] [CrossRef] [PubMed]
- Hajda, J.; Lehmann, M.; Krebs, O.; Kieser, M.; Geletneky, K.; Jager, D.; Dahm, M.; Huber, B.; Schoning, T.; Sedlaczek, O.; et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer 2017, 17, 576. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Mutation(s) | Modified Protein(s) | Modified Amino Acid(s) |
|---|---|---|---|
| H-1-PM-I | T2044C | NS1/NS2 | Tyr595His/Leu103Pro |
| H-1-PM-II | A2022G | NS2 | Lys96Glu |
| H-1-DM | T2044C, A2022G | NS1/NS2 | Tyr595His/Leu103Pro, Lys96Glu |
| H-1-PM-III | C2193A | NS2 | Leu153Met |
| Del-H-1PV | Δnt2022-2135 | NS1/NS2 | 38 aa deleted |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashemi, H.; Condurat, A.-L.; Stroh-Dege, A.; Weiss, N.; Geiss, C.; Pilet, J.; Cornet Bartolomé, C.; Rommelaere, J.; Salomé, N.; Dinsart, C. Mutations in the Non-Structural Protein-Coding Sequence of Protoparvovirus H-1PV Enhance the Fitness of the Virus and Show Key Benefits Regarding the Transduction Efficiency of Derived Vectors. Viruses 2018, 10, 150. https://doi.org/10.3390/v10040150
Hashemi H, Condurat A-L, Stroh-Dege A, Weiss N, Geiss C, Pilet J, Cornet Bartolomé C, Rommelaere J, Salomé N, Dinsart C. Mutations in the Non-Structural Protein-Coding Sequence of Protoparvovirus H-1PV Enhance the Fitness of the Virus and Show Key Benefits Regarding the Transduction Efficiency of Derived Vectors. Viruses. 2018; 10(4):150. https://doi.org/10.3390/v10040150
Chicago/Turabian StyleHashemi, Hamidreza, Alexandra-Larisa Condurat, Alexandra Stroh-Dege, Nadine Weiss, Carsten Geiss, Jill Pilet, Carles Cornet Bartolomé, Jean Rommelaere, Nathalie Salomé, and Christiane Dinsart. 2018. "Mutations in the Non-Structural Protein-Coding Sequence of Protoparvovirus H-1PV Enhance the Fitness of the Virus and Show Key Benefits Regarding the Transduction Efficiency of Derived Vectors" Viruses 10, no. 4: 150. https://doi.org/10.3390/v10040150