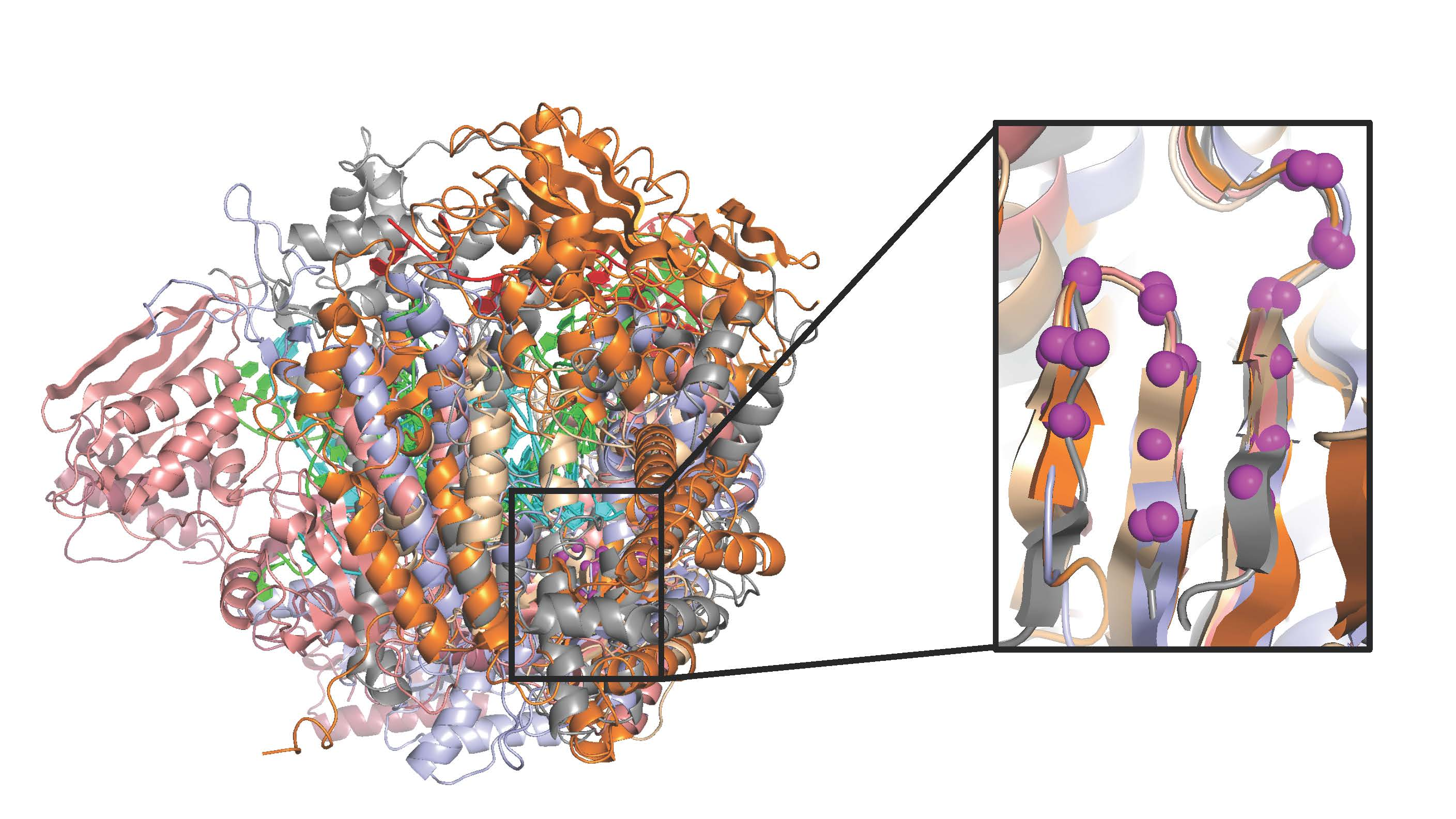
There are two fundamental issues for generating reliable structure superpositions; first, identifying the residues that should be considered structurally equivalent, and second, reorienting all the individual structures to optimally align these matched residues with each other. While it is straightforward to visually appreciate that viral polymerases share the core conserved active site motifs, it is somewhat more complicated to develop a comprehensive superpositioning scheme that would allow comparisons across all the structures solved to date. One could pick a “master” polymerase and pairwise align all other polymerases to this, but such a comparison would be limited to a rather small region of strong structural similarity. A better approach would be to use those parts of the entire structures that share the same fold. This means the superposed regions could become larger when the structural similarity is greater, and in the extreme case of comparing many structures of the same polymerase it would be best to use complete structures. For the more common case of somewhat divergent polymerases it would be best to use only their structurally conserved regions, for example the core motifs A, B, and C that surround the active site. However, a major downside of this approach is that it requires a meticulous one-to-one mapping of structurally equivalent residues, which becomes more difficult as structures diverge. In fact, the diversity among all viral polymerases means that only a small portion of their structures within motifs A and C could be considered truly similar for such a global comparison, as illustrated by
Figure 2 and
Figure S2.
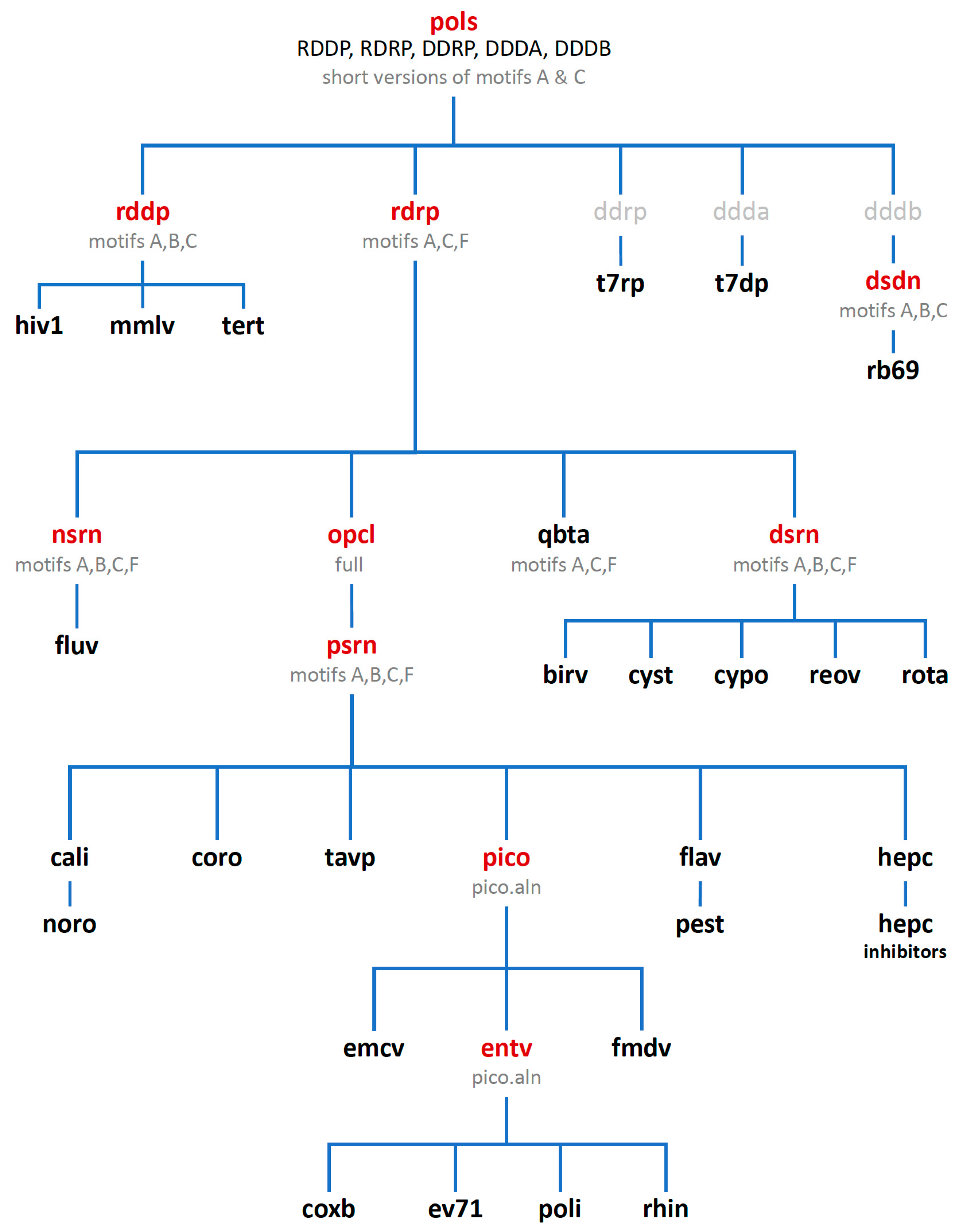
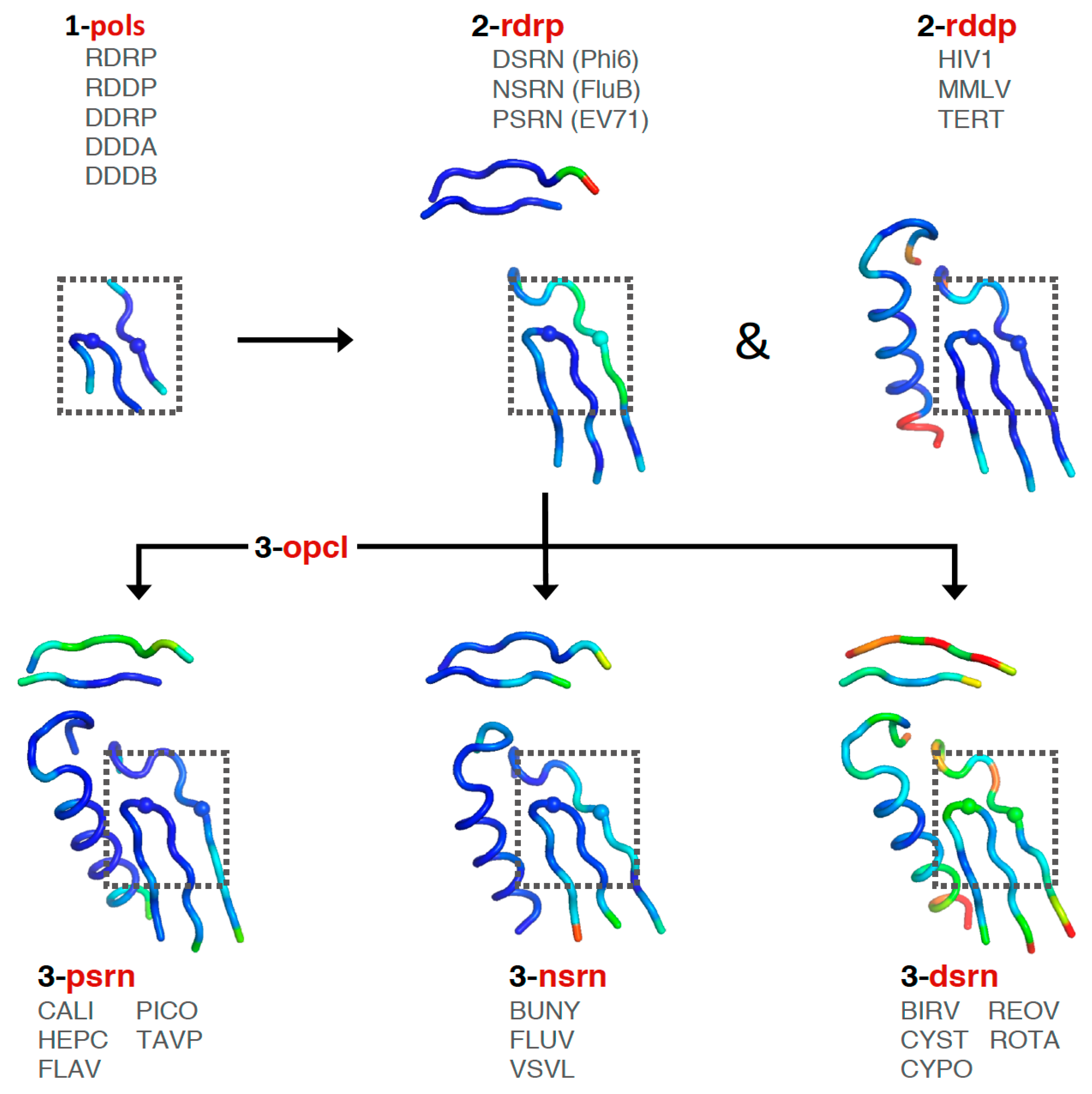
Consideration of these factors led to the development of a tree-based organization for polymerase structure alignments wherein each node on the tree is a multiple structure alignment that inherits its orientation from the previous node on the tree. The overall tree is illustrated in
Figure 3, where the nodes have four-letter names corresponding to alignment “
sets” that will be indicated in boldface throughout the text. The tree starts with the
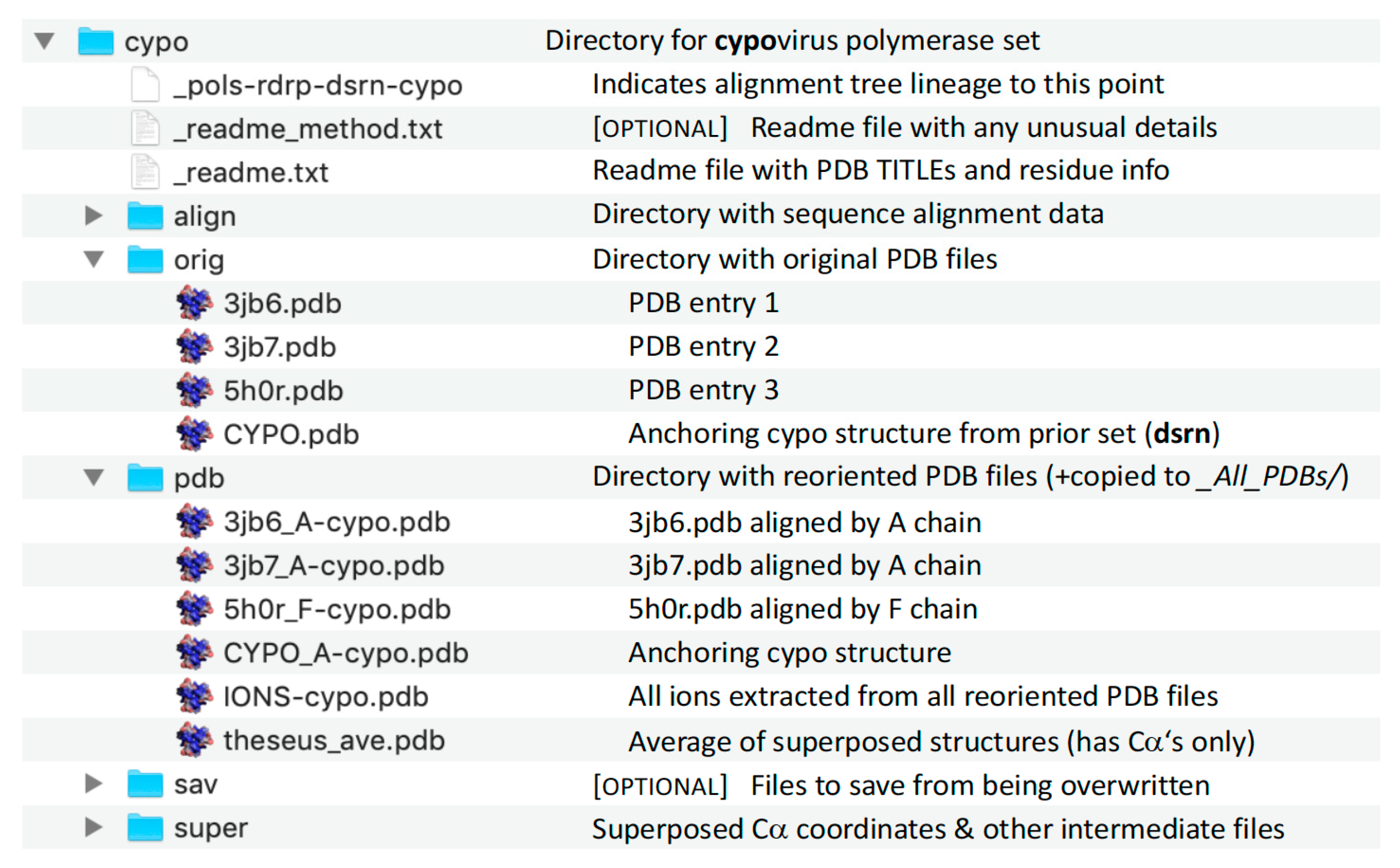
pols set that is a superpositioning of minimal shared core segments from the four major classes of DNA- or RNA-dependent DNA or RNA polymerases, i.e., DdDP, DdRP, RdDP, and RdRP enzymes. Two different DdDP structures were included, one from an A family polymerase (DDDA, T7 DNA polymerase) and one from a B family polymerase (DDDB, RB69 polymerase). The orientations of the five structures resulting from this alignment are then retained as each of them seeds one of the next sets that superpose more closely related polymerases. These have larger regions of similarity, which will expand and improve the subsequent alignments. For example, the RdDP structure from the
pols set becomes the master orientation for the subsequent
rddp set that superposes reverse transcriptase structures from HIV, Moloney murine leukemia virus (
mmlv), and telomerase. This process is repeated as necessary to build out the tree, with the branches ending in superpositions of either very similar proteins, such as the
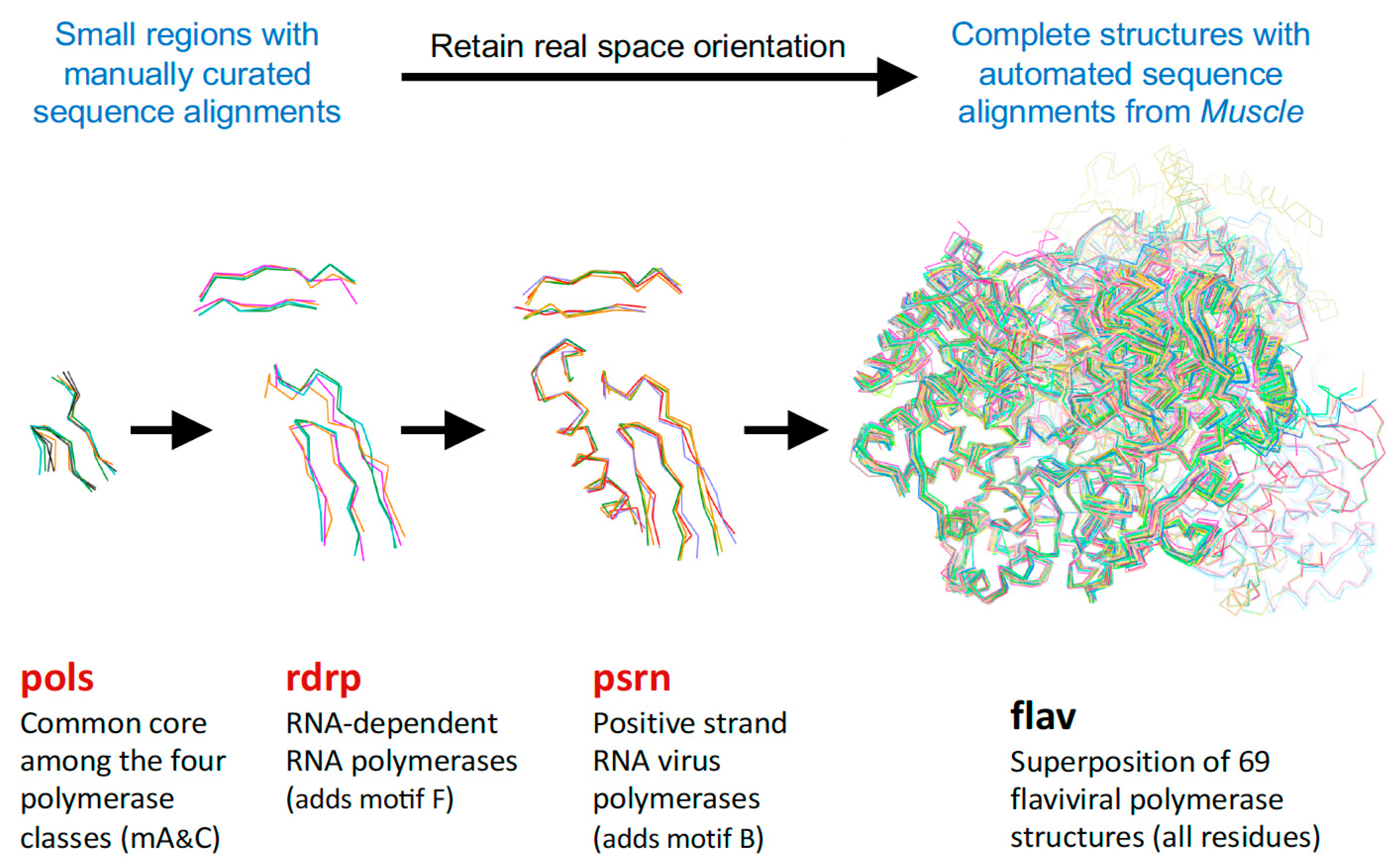
flav set with all flaviviral polymerase structures, or multiple structures of the same protein, such as the
ev71 set with all available enterovirus 71 polymerase structures. Each superposition set named in
Figure 3 corresponds to a single multiple structure alignment, and comparisons of structures within a single set will be the most reliable. But the consistent mapping back to the parental orientation in the tree means that one can mix and match PDB coordinate files from distantly related branches and retain fairly reliable alignment. The overall result is 646 different coordinate files corresponding to 646 different polymerase chains taken from 414 different original PDB files, all of which have been reoriented into a common coordinate space.
3.1. Maximum Likelihood Superpositions with Theseus
The second major issue is the mathematical treatment used for the alignment itself, i.e., how does one determine the reorientation matrix that is used to move a structure from its original orientation into a new orientation where it is superimposed on another structure. For this I chose to use maximum likelihood based multiple structure alignments with the program Theseus [
4,
5,
6]. Unlike a traditional pairwise least-squares alignment of two structures where every atom position is weighted equally, the maximum likelihood method simultaneously aligns multiple structures and considers the statistical distributions of coordinates at every position to arrive at a globally optimal fit. Without going into the mathematical details, which are well described in [
7], the end result is to effectively down weight structurally divergent regions and converge about the most structurally similar regions. This effect can be appreciated by the superposition comparison figure shown in [
5] and on the
www.theseus3d.org web site, where a family of NMR structures is used to show that the maximum likelihood (ML) alignment is clearly superior to the classical least-squares (LS) alignment that is biased by the conformational heterogeneity in internal loops and the ends of the protein.
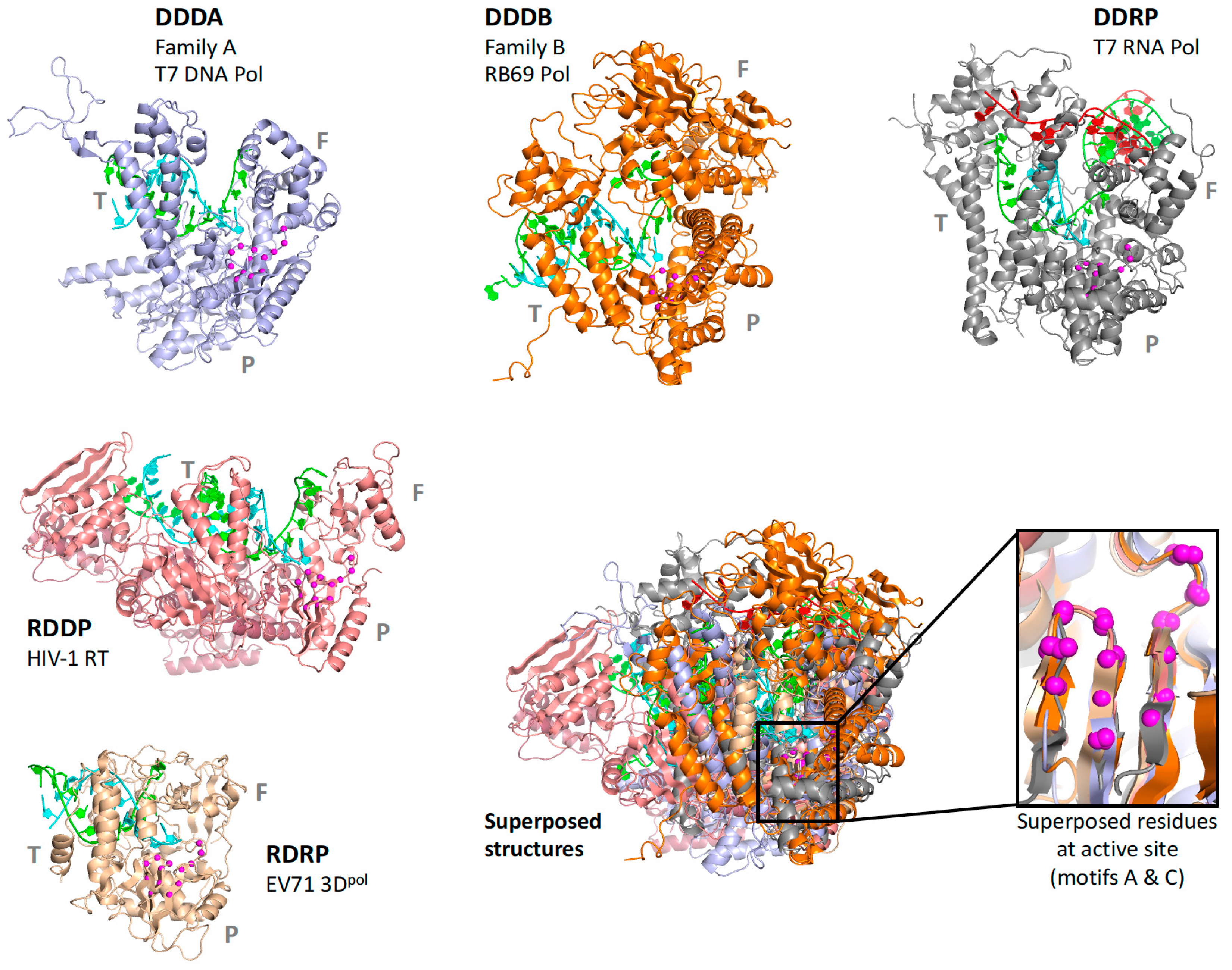
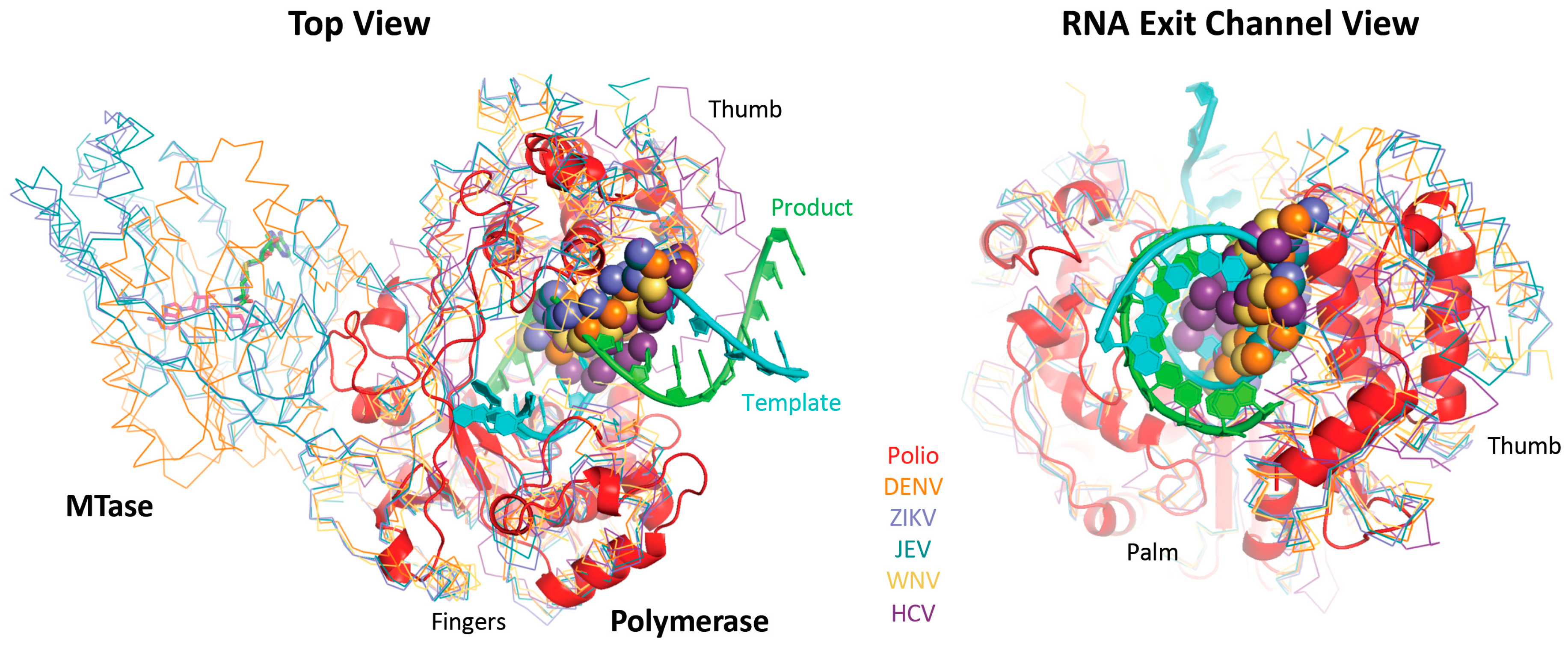
For the polymerase superposition tree, the branch termini are usually groups of identical or closely related polymerases with tight distributions of conformations because the structures are fairly similar, e.g.,
flav set with NS5 structures from dengue, West Nile, zika, and Japanese encephalitis viruses as shown in
Figure 4. Within such groups the similarities in both sequence and structure space are high enough that the full-structure superpositioning process can be automated by the method outlined in
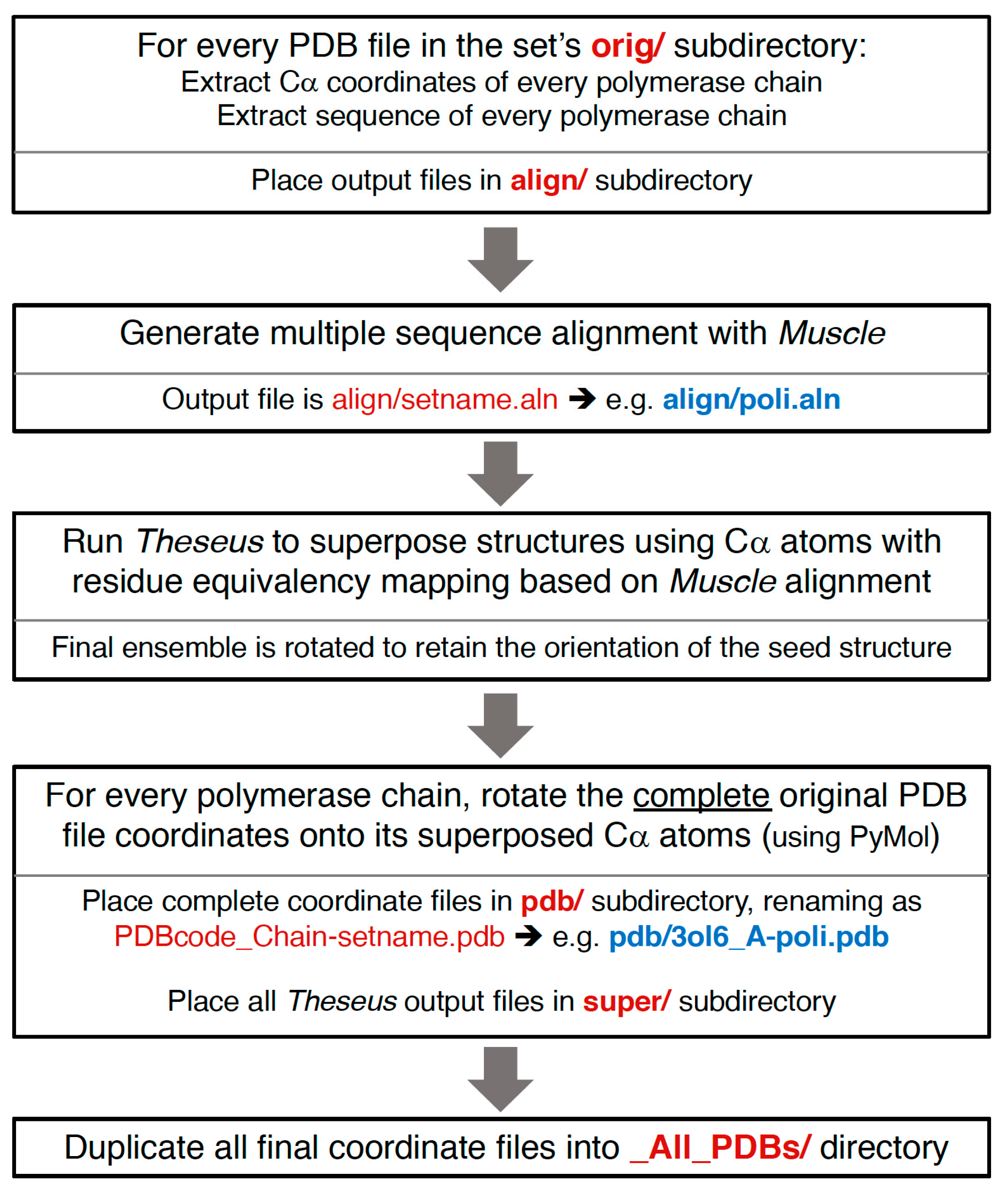
Section 4. This basically consists of assigning equivalent residues via a multiple sequence alignment from Muscle [
8] and then using Theseus to align the structures in three dimensions based on Cα positions only. Notably, while minor errors in the sequence alignment can result in mis-assigning structurally equivalent residues, this does not significantly affect the final superposition because such positions will mathematically appear as divergent parts of the structure and consequently be effectively down-weighted in the analysis.
3.2. Picornaviral Polymerases as an Example
Figure 5,
Figure 6, and
Figure S3 further illustrate the structure superposition hierarchy starting with the
pols set and ending with
poli that contains all solved poliovirus polymerase structures. Whereas
Figure 4 showed the individual structures for each step in the hierarchy,
Figure 5 shows the average structure as a cartoon “worm” with spheres for the Cα atoms of the two conserved active site aspartate residues in motifs A and C. To give a visual indication of structural heterogeneity across the superposed region, the worm is colored according to a pseudo B-factor calculated as (8π
2) times the statistical variance observed at each Cα position. The representative structures chosen for each set contained a canonical conformation across the superposed region so as not to bias the resulting orientations.
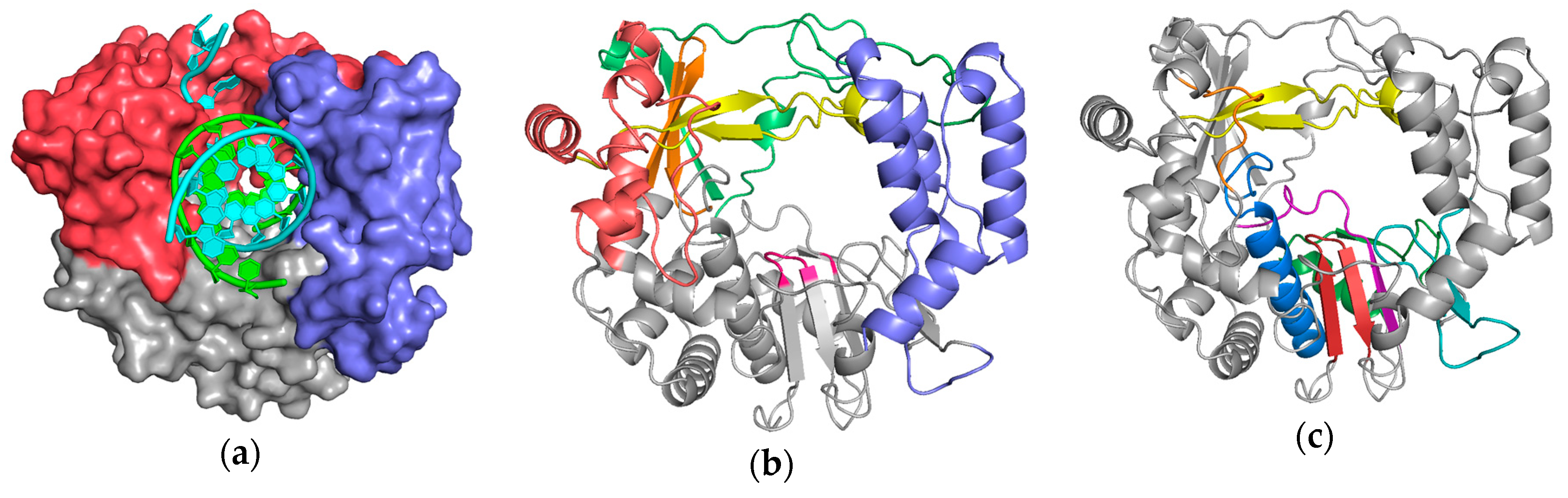
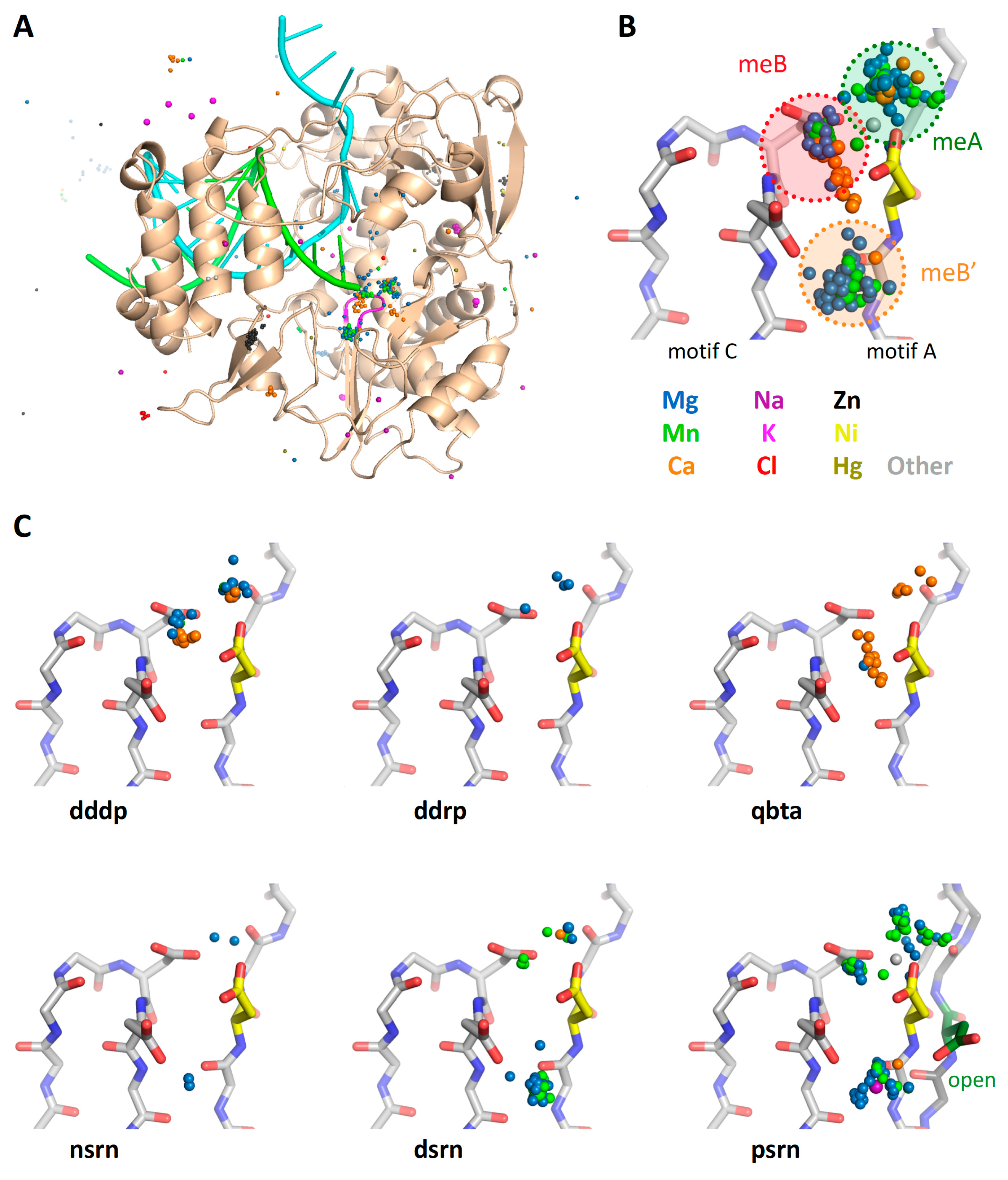
The first stage superposition set (
pols) provides the base orientations for representative structures of the four classes of polymerases, and it is done using minimal 13-residue versions of the core motifs A and C that represent the limited extent of active site structural similarity among these highly divergent polymerases. The RdRP structure from this superposition is then used to anchor the orientation of the
rdrp set consisting of three different viral RdRPs—one each from the positive strand (
psrn), negative strand (
nsrn), and double stranded RNA (
dsrn) viruses. This
rdrp set expands motifs A and C from 13 to 26 residues and adds 14 residues from motif F, the pair of antiparallel β-strands found above the active site in RdRPs, resulting in a total of 40 residues (also shown in
Figure 4). The subsequent
nsrn set was expanded with an influenza virus specific set and the
dsrn set was further expanded with five sets corresponding to different dsRNA viruses (see
Figure 2). Bacteriophage Qβ was given its own
qbta set by superimposing all Qβ replicase structures onto core motifs A, C, and F from the representative RdRP structure used in the
pols set.
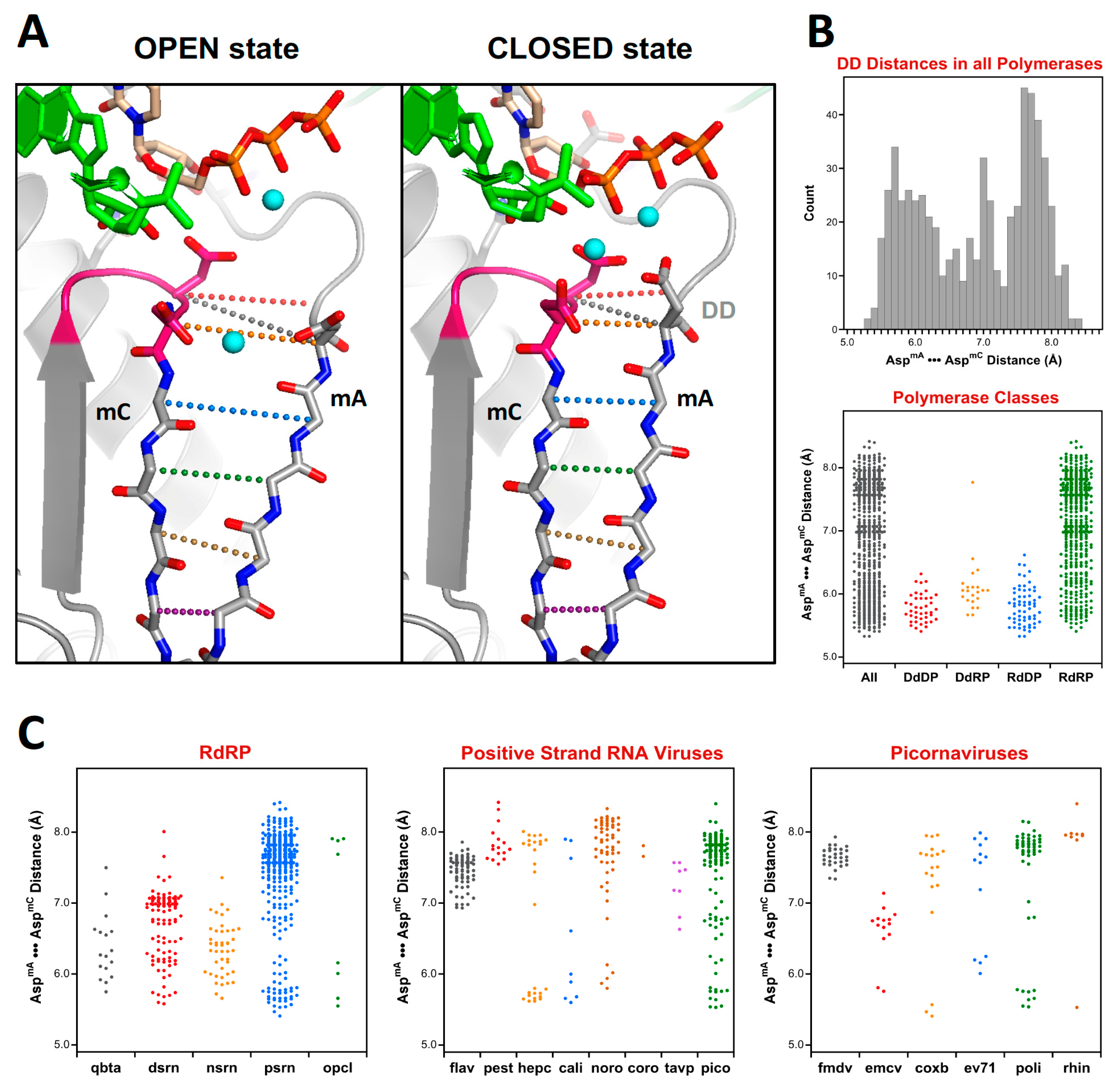
At this point there is a significant divergence in the active site structures of the positive strand RNA virus (
psrn) polymerases compared to all the other groups of polymerases. This arises because these enzymes close their active sites for catalysis by a subtle movement of motif A that completes the β-sheet between motifs A and C [
9]. The default structures of most
psrn RdRPs, solved in the absence of RNA and NTPs, reflect the open state of the active site. However, several of these RdRPs have been captured in the closed state when crystallized as elongation complexes with RNA and a correct nucleotide triphosphate. While the differences between the two states are structurally subtle, as described in
Section 5.6, they are significant at the level of active site superpositions, particularly in the initial
pols and
rdrp sets where the core active site structures of other polymerase classes reflect the closed state of a
psrn RdRP. To accommodate this structural transition in the alignment tree, a special superposition set called
opcl was devised to interconvert between the open conformation that is predominant among the positive strand virus RdRP structures and the closed conformation found in the prior alignment sets (
pols and
rdrp) of the tree. To limit bias from any one pair of open versus closed structures, this
opcl set was assembled from eight different structures that represent two pairs each of poliovirus and enterovirus 71 3D
pol structures that were solved in both the absence and presence of NTP by soaking experiments of identical crystals [
9,
10]. The complete structures were used such that the conformational changes associated with the open vs. closed active sites did not significantly affect the alignment. The orientation inherited from the
rdrp set is a closed form EV71 polymerase structure (PDB: 5F8J) and the corresponding open form structure (PDB: 5F8G) is used as the seed orientation for the subsequent alignment of all positive-strand RNA polymerases.
The next
psrn set is a superposition of six different positive-strand RNA virus polymerases and the aligned region has been expanded from 40 to 65 residues by including motif B that forms a loop structure followed by a long α-helix that lies adjacent to motifs A and C. The representative structures in this
psrn alignment in turn define the parent orientations for subsequent alignments of caliciviral (
cali), coronaviral (
coro), flaviviral (
flav), hepatitis C (
hepc), picornaviral (
pico), and
Thosea asigna virus (
tavp) polymerases. A set for the noroviruses (
noro) was generated under the
cali set, and a pestivirus set (
pest) was made under the
flav set. The
pest set used the representative flaviviral structure (FLAV.pdb) from
psrn as an alignment seed to avoid over-representing flaviviral polymerases in the
psrn set. Note that
Thosea asigna virus polymerase has an altered topology where motif C precedes motif A in the primary sequence, but the active site conformation is conserved nonetheless. There are interesting structural parallels between this and the double stranded RNA birnavirus (
birv) polymerase that shares this non-canonical topology [
11].
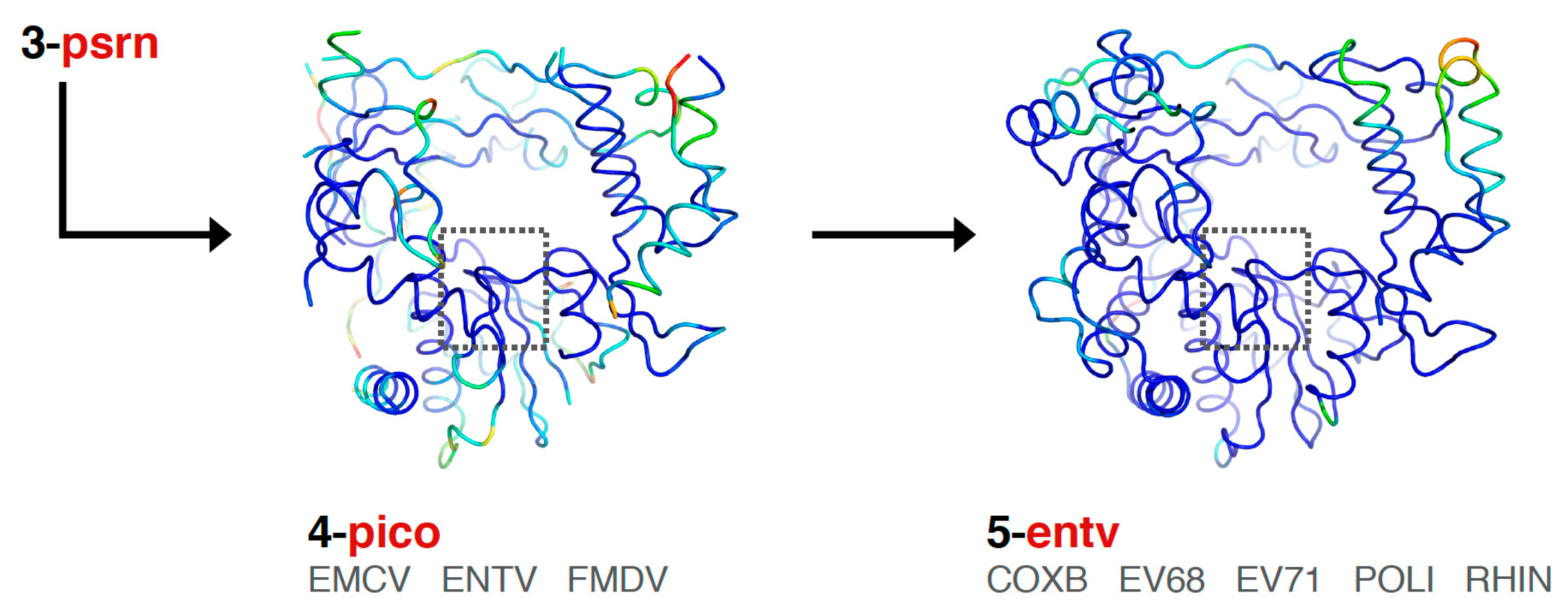
Finally, the picornaviral polymerases are aligned in two stages that end in sets superposing all available structures of each viral polymerase (
Figure 6). First, the
pico set is composed of three structures representing encephalomyocarditis virus (
emcv), foot-and-mouth disease virus (
fmdv), and poliovirus 3D
pol as a representative enterovirus (
entv). This was done because there is a difference in the orientation of a pinky finger helix for these structures, with FMDV and EMCV being similar to each other yet distinct from the enteroviruses (
Figure S3, scene F5). The second stage is an
entv set with six polymerases that give rise to separate coxsackievirus B (
coxb), enterovirus 71 (
ev71), poliovirus (
poli), and rhinovirus (
rhin) alignment sets. Each of these enterovirus sets in turn superimpose all available structures of each polymerase. The picornaviral polymerase alignments are done using the complete ≈460-residue structures, not just the core motifs as for previous sets in the tree, and they are based on a manually curated structure-based sequence alignment (the file 4-pico/sav/pico_v5.aln,
Figure S5). This alignment assigns all structurally equivalent residues and accounts for both residue insertions/deletions and regions with significantly different conformations, e.g., the pinky finger helix mentioned above, by making non-overlapping sections in the sequence alignment so that the structure alignment does not attempt to superpose these divergent structures on each other. The single available EV-D68 structure (PDB: 5XE0) was included in the
entv set, and the single available coxsackievirus A16 structure (PDB: 5Y6Z) was included in the
ev71 set.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











