
Identification of Multiple Novel Viruses in Fecal Samples of Black-Necked Cranes Using Viral Metagenomic Methods
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Viral Nucleic Acid Extraction
2.3. Library Construction and Bioinformatics Analysis
2.4. Phylogenetic Analysis
2.5. Sequence Alignment and ORF Prediction
2.6. Nucleotide Sequence Accession Number
3. Results
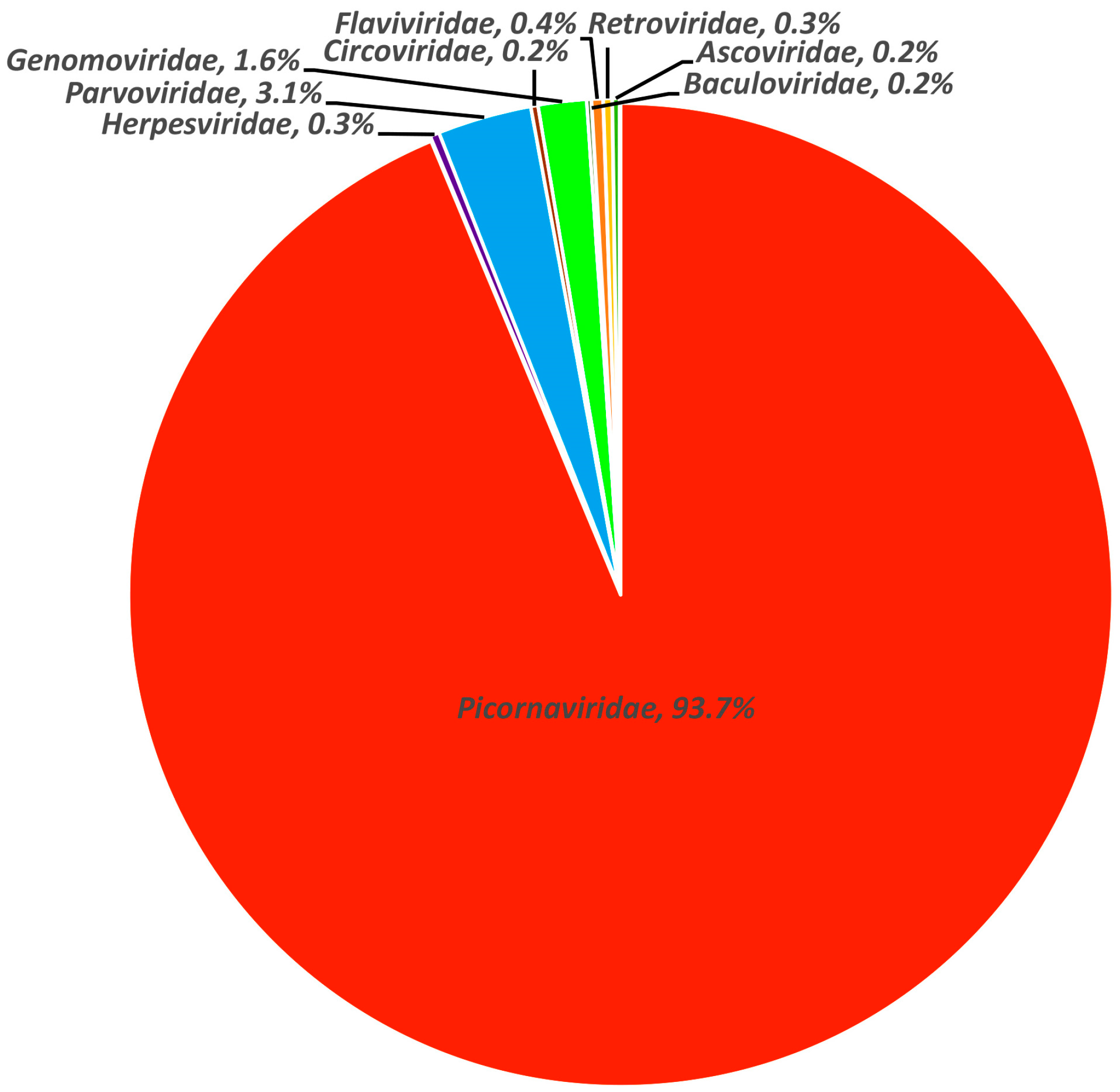
3.1. Viral Metagenomic Overview
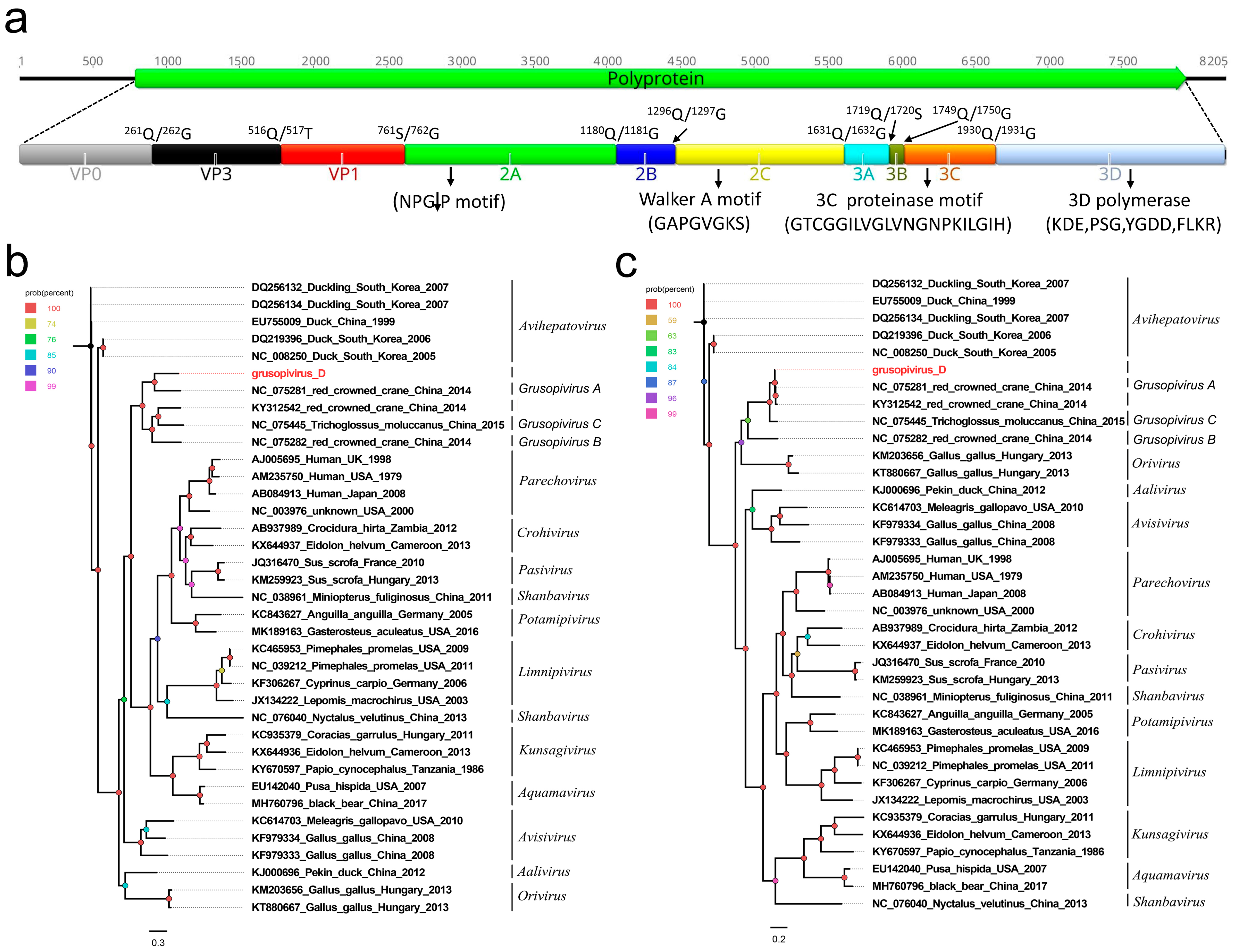
3.2. A Novel Picornavirus Belonging to the Genus Grusopivirus of the Family Picornaviridae
3.3. Four New Parvoviruses Belonging to the Genus Chaphamaparvovirus of the Family Parvoviridae
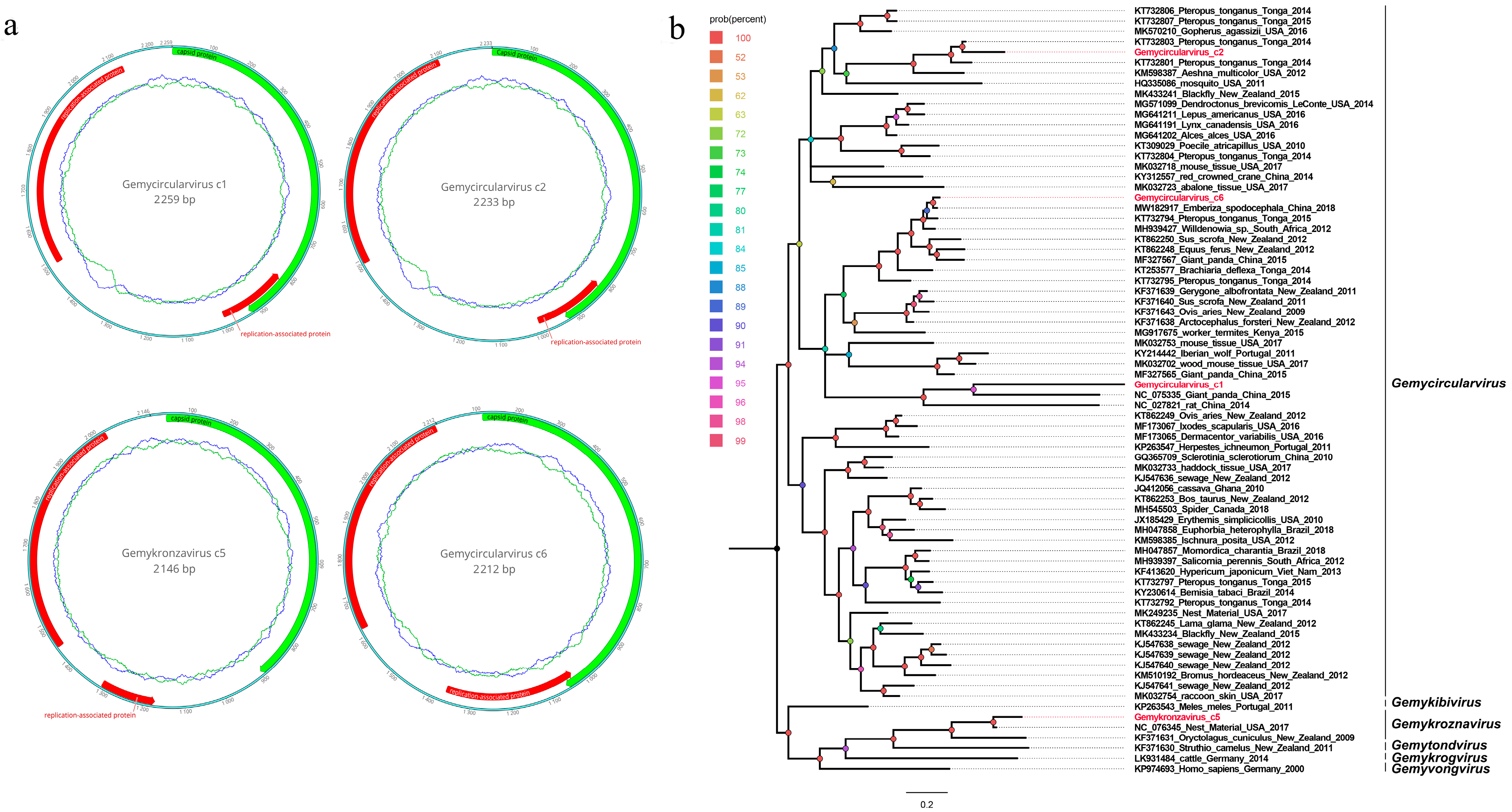
3.4. Four Novel Genomoviruses Belonging to the Family Genomoviidae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- BirdLife IUCN Red List of Threatened Species: Grus Nigricollis. Available online: https://www.iucnredlist.org/species/22692162/180030167#assessment-information (accessed on 24 July 2020).
- Yang, Y.; Xie, C.; Shen, C.; Tian, B.; Wang, S.; Bian, X.; Guo, Y.; Zhu, Y.; Fang, H. Changes in the Landscape Patterns of Black-Necked Crane Habitat and Its Correlation with Their Individual Population Numbers during the Past 40 Years in China. Ecol. Evol. 2023, 13, e10125. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yu, H.; Geng, Y.; Liu, W.; Zheng, S.; Yang, N.; Meng, Y.; Dou, L.; Price, M.; Ran, J.; et al. A High-Quality Draft Genome Assembly of the Black-Necked Crane (Grus nigricollis) Based on Nanopore Sequencing. Genome Biol. Evol. 2019, 11, 3332–3340. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, Y.; DuBay, S.G.; Zhao, C.; Wang, B.; Ran, J. Dung-Associated Arthropods Influence Foraging Ecology and Habitat Selection in Black-Necked Cranes (Grus nigricollis) on the Qinghai-Tibet Plateau. Ecol. Evol. 2019, 9, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.-J.; Lee, Y.-N.; Cheon, S.-H.; Park, Y.-R.; Baek, Y.-G.; Kye, S.-J.; Lee, M.-H.; Lee, Y.-J. Isolation and Characterization of Low Pathogenic H7N7 Avian Influenza Virus from a Red-Crowned Crane in a Zoo in South Korea. BMC Vet. Res. 2020, 16, 432. [Google Scholar] [CrossRef]
- Olsen, G.H.; Miller, K.J.; Docherty, D.E.; Bochsler, V.S.; Sileo, L. Pathogenicity of West Nile Virus and Response to Vaccination in Sandhill Cranes (Grus canadensis) Using a Killed Vaccine. J. Zoo Wildl. Med. 2009, 40, 263–271. [Google Scholar] [CrossRef]
- Lian, X.; Ming, X.; Xu, J.; Cheng, W.; Zhang, X.; Chen, H.; Ding, C.; Jung, Y.-S.; Qian, Y. First Molecular Detection and Characterization of Marek’s Disease Virus in Red-Crowned Cranes (Grus japonensis): A Case Report. BMC Vet. Res. 2018, 14, 122. [Google Scholar] [CrossRef]
- Foerster, S.; Chastel, C.; Kaleta, E.F. Crane Hepatitis Herpesviruses. Zentralbl. Veterinarmed. B 1989, 36, 433–441. [Google Scholar] [CrossRef]
- Shan, T.; Yang, S.; Wang, H.; Wang, H.; Zhang, J.; Gong, G.; Xiao, Y.; Yang, J.; Wang, X.; Lu, J.; et al. Virome in the Cloaca of Wild and Breeding Birds Revealed a Diversity of Significant Viruses. Microbiome 2022, 10, 60. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, D.; Ji, Z.; Zhang, Y.; Wang, Y.; Chen, X.; He, Y.; Lu, X.; Li, R.; Guo, Y.; et al. Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs. Viruses 2022, 14, 2048. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The Fecal Virome of Red-Crowned Cranes. Arch. Virol. 2019, 164, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y.; Li, X.; Chen, R.; Li, W.; Ji, L.; Zhao, Q.; Ji, L.; Yang, S.; Zhang, W. Molecular Detection and Characterization of Three Novel Parvoviruses Belonging to Two Different Subfamilies in Zoo Birds. Arch. Virol. 2023, 168, 163. [Google Scholar] [CrossRef]
- Xiao, C.-T.; Giménez-Lirola, L.G.; Jiang, Y.-H.; Halbur, P.G.; Opriessnig, T. Characterization of a Novel Porcine Parvovirus Tentatively Designated PPV5. PLoS ONE 2013, 8, e65312. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Male, M.F.; Kraberger, S.; Stainton, D.; Kami, V.; Varsani, A. Cycloviruses, Gemycircularviruses and Other Novel Replication-Associated Protein Encoding Circular Viruses in Pacific Flying Fox (Pteropus tonganus) Faeces. Infect. Genet. Evol. 2016, 39, 279–292. [Google Scholar] [CrossRef]
- Dai, Z.; Wang, H.; Wu, H.; Zhang, Q.; Ji, L.; Wang, X.; Shen, Q.; Yang, S.; Ma, X.; Shan, T.; et al. Parvovirus Dark Matter in the Cloaca of Wild Birds. Gigascience 2022, 12, giad001. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, S.; Shan, T.; Hou, R.; Liu, Z.; Li, W.; Guo, L.; Wang, Y.; Chen, P.; Wang, X.; et al. Virome Comparisons in Wild-Diseased and Healthy Captive Giant Pandas. Microbiome 2017, 5, 90. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Family Genomoviridae: 2021 Taxonomy Update. Arch. Virol. 2021, 166, 2911–2926. [Google Scholar] [CrossRef]
- Esaki, M.; Ito, G.; Tokorozaki, K.; Matsui, T.; Masatani, T.; Amano, K.; Ozawa, M. Prevalence and Organ Tropism of Crane-Associated Adenovirus 1 in Cranes Overwintering on the Izumi Plain, Japan. Transbound. Emerg. Dis. 2022, 69, e2800–e2807. [Google Scholar] [CrossRef]
- Uwatoko, K.; Inaba, Y.; Sasaki, T.; Sano, Y.; Pan, I.J.; Suzuki, H.; Yamaguchi, S.; Taniguchi, T. Hemagglutination with Crane Herpesvirus. J. Vet. Med. Sci. 1998, 60, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Knowles, N.J.; Simmonds, P. A Proposed Division of the Family Picornaviridae into Subfamilies Based on Phylogenetic Relationships and Functional Genomic Organization. Arch. Virol. 2021, 166, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Wang, Y.; Yao, X.; Mao, Q.; Xu, M.; Liang, Z. Coxsackievirus A6: A New Emerging Pathogen Causing Hand, Foot and Mouth Disease Outbreaks Worldwide. Expert Rev. Anti Infect. Ther. 2015, 13, 1061–1071. [Google Scholar] [CrossRef]
- Huaman, J.L.; Pacioni, C.; Sarker, S.; Doyle, M.; Forsyth, D.M.; Pople, A.; Carvalho, T.G.; Helbig, K.J. Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia. Viruses 2021, 13, 2412. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Zell, R.; Deboutte, W.; Zeller, M.; Conceição-Neto, N.; Heylen, E.; Maes, P.; Knowles, N.J.; Ghogomu, S.M.; Van Ranst, M.; et al. Highly Diverse Population of Picornaviridae and Other Members of the Picornavirales, in Cameroonian Fruit Bats. BMC Genom. 2017, 18, 249. [Google Scholar] [CrossRef] [PubMed]
- Philipps, A.; Dauber, M.; Groth, M.; Schirrmeier, H.; Platzer, M.; Krumbholz, A.; Wutzler, P.; Zell, R. Isolation and Molecular Characterization of a Second Serotype of the Encephalomyocarditis Virus. Vet. Microbiol. 2012, 161, 49–57. [Google Scholar] [CrossRef]
- Pankovics, P.; Boros, Á.; Kiss, T.; Reuter, G. Identification and Complete Genome Analysis of Kobuvirus in Faecal Samples of European Roller (Coracias garrulus): For the First Time in a Bird. Arch. Virol. 2015, 160, 345–351. [Google Scholar] [CrossRef]
- Ramirez-Fort, M.K.; Downing, C.; Doan, H.Q.; Benoist, F.; Oberste, M.S.; Khan, F.; Tyring, S.K. Coxsackievirus A6 Associated Hand, Foot and Mouth Disease in Adults: Clinical Presentation and Review of the Literature. J. Clin. Virol. 2014, 60, 381–386. [Google Scholar] [CrossRef]
- Uprety, P.; Graf, E.H. Enterovirus Infection and Acute Flaccid Myelitis. Curr. Opin. Virol. 2020, 40, 55–60. [Google Scholar] [CrossRef]
- Song, J.; Lu, H.; Ma, L.; Zhu, S.; Yan, D.; Han, J.; Zhang, Y. Molecular Characteristics of Enterovirus B83 Strain Isolated from a Patient with Acute Viral Myocarditis and Global Transmission Dynamics. Viruses 2023, 15, 1360. [Google Scholar] [CrossRef]
- Bauer, D.; Farthofer, A.; Chromy, D.; Simbrunner, B.; Steininger, L.; Schmidbauer, C.; Binter, T.; Trauner, M.; Mandorfer, M.; Schmidt, R.; et al. Recent Outbreaks of Severe Hepatitis A Virus Infections in Vienna. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Montiel, N.; Buckley, A.; Guo, B.; Kulshreshtha, V.; VanGeelen, A.; Hoang, H.; Rademacher, C.; Yoon, K.-J.; Lager, K. Vesicular Disease in 9-Week-Old Pigs Experimentally Infected with Senecavirus A. Emerg. Infect. Dis. 2016, 22, 1246–1248. [Google Scholar] [CrossRef] [PubMed]
- Mietzsch, M.; Pénzes, J.J.; Agbandje-McKenna, M. Twenty-Five Years of Structural Parvovirology. Viruses 2019, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An Ancient Lineage of Highly Divergent Parvoviruses Infects Both Vertebrate and Invertebrate Hosts. Viruses 2019, 11, 525. [Google Scholar] [CrossRef]
- Palombieri, A.; Di Profio, F.; Lanave, G.; Capozza, P.; Marsilio, F.; Martella, V.; Di Martino, B. Molecular Detection and Characterization of Carnivore Chaphamaparvovirus 1 in Dogs. Vet. Microbiol. 2020, 251, 108878. [Google Scholar] [CrossRef]
- Cui, H.; Pan, S.; Xu, X.; Ji, J.; Ma, K.; Yao, L.; Kan, Y.; Bi, Y.; Xie, Q. Molecular Characteristics of Novel Chaphamaparvovirus Identified in Chickens. Poult. Sci. 2023, 102, 102449. [Google Scholar] [CrossRef]
- Matos, M.; Bilic, I.; Viloux, N.; Palmieri, N.; Albaric, O.; Chatenet, X.; Tvarogová, J.; Dinhopl, N.; Heidl, S.; Liebhart, D.; et al. A Novel Chaphamaparvovirus Is the Etiological Agent of Hepatitis Outbreaks in Pheasants (Phasianus colchicus) Characterized by High Mortality. Transbound. Emerg. Dis. 2022, 69, e2093–e2104. [Google Scholar] [CrossRef]
- Ramos, E.d.S.F.; Abreu, W.U.; Rodrigues, L.R.R.; Marinho, L.F.; Morais, V.D.S.; Villanova, F.; Pandey, R.P.; Araújo, E.L.L.; Deng, X.; Delwart, E.; et al. Novel Chaphamaparvovirus in Insectivorous Molossus Molossus Bats, from the Brazilian Amazon Region. Viruses 2023, 15, 606. [Google Scholar] [CrossRef]
- Sarker, S. Molecular and Phylogenetic Characterisation of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots. Pathogens 2021, 10, 1559. [Google Scholar] [CrossRef]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.A.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An Atypical Parvovirus Drives Chronic Tubulointerstitial Nephropathy and Kidney Fibrosis. Cell 2018, 175, 530–543. [Google Scholar] [CrossRef]
- Sarker, S. Characterization of a Novel Complete-Genome Sequence of a Galliform Chaphamaparvovirus from a Free-Range Laying Chicken Clinically Diagnosed with Spotty Liver Disease. Microbiol. Resour. Announc. 2022, 11, e0101722. [Google Scholar] [CrossRef] [PubMed]
- Fahsbender, E.; Altan, E.; Seguin, M.A.; Young, P.; Estrada, M.; Leutenegger, C.; Delwart, E. Chapparvovirus DNA Found in 4% of Dogs with Diarrhea. Viruses 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ghabrial, S.A.; Jiang, D.; Varsani, A. Genomoviridae: A New Family of Widespread Single-Stranded DNA Viruses. Arch. Virol. 2016, 161, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Tang, Y.; Zhang, S.; She, X.; Lan, G.; Varsani, A.; He, Z. Identification and Molecular Characterization of a Single-Stranded Circular DNA Virus with Similarities to Sclerotinia Sclerotiorum Hypovirulence-Associated DNA Virus 1. Arch. Virol. 2014, 159, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gu, Y.; Shen, Q.; Yang, S.; Wang, X.; Wan, Y.; Zhang, W. A Novel Gemycircularvirus from Experimental Rats. Virus Genes 2015, 51, 302–305. [Google Scholar] [CrossRef] [PubMed]
- da Silva Assis, M.R.; Vieira, C.B.; Fioretti, J.M.; Rocha, M.S.; de Almeida, P.I.N.; Miagostovich, M.P.; Fumian, T.M. Detection and Molecular Characterization of Gemycircularvirus from Environmental Samples in Brazil. Food Environ. Virol. 2016, 8, 305–309. [Google Scholar] [CrossRef]
- Schmidlin, K.; Sepp, T.; Khalifeh, A.; Smith, K.; Fontenele, R.S.; McGraw, K.J.; Varsani, A. Diverse Genomoviruses Representing Eight New and One Known Species Identified in Feces and Nests of House Finches (Haemorhous mexicanus). Arch. Virol. 2019, 164, 2345–2350. [Google Scholar] [CrossRef]
- Hanna, Z.R.; Runckel, C.; Fuchs, J.; DeRisi, J.L.; Mindell, D.P.; Van Hemert, C.; Handel, C.M.; Dumbacher, J.P. Isolation of a Complete Circular Virus Genome Sequence from an Alaskan Black-Capped Chickadee (Poecile atricapillus) Gastrointestinal Tract Sample. Genome Announc. 2015, 3, 207:1–207:2. [Google Scholar] [CrossRef]
- Chiumenti, M.; Greco, C.; Antelmi, I.; Sion, V.; Altamura, G.; Nigro, F.; Saldarelli, P. Molecular Characterisation of a Novel Gemycircularvirus Associated with Olive Trees in Italy. Virus Res. 2019, 263, 169–172. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Zhao, R.; Sun, Y.; Ji, L.; Xi, Y.; Wang, X.; Shen, Q.; Ji, L.; Wang, Y.; You, Z.; et al. Identification of Multiple Novel Viruses in Fecal Samples of Black-Necked Cranes Using Viral Metagenomic Methods. Viruses 2023, 15, 2068. https://doi.org/10.3390/v15102068
Zhao Q, Zhao R, Sun Y, Ji L, Xi Y, Wang X, Shen Q, Ji L, Wang Y, You Z, et al. Identification of Multiple Novel Viruses in Fecal Samples of Black-Necked Cranes Using Viral Metagenomic Methods. Viruses. 2023; 15(10):2068. https://doi.org/10.3390/v15102068
Chicago/Turabian StyleZhao, Qifan, Ran Zhao, Yijie Sun, Li Ji, Yuan Xi, Xiaochun Wang, Quan Shen, Likai Ji, Yan Wang, Zhenqiang You, and et al. 2023. "Identification of Multiple Novel Viruses in Fecal Samples of Black-Necked Cranes Using Viral Metagenomic Methods" Viruses 15, no. 10: 2068. https://doi.org/10.3390/v15102068