
Epidemiological Features of Human Norovirus Genotypes before and after COVID-19 Countermeasures in Osaka, Japan

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Detection of HuNoV
2.3. Sequencing and Genotyping
2.4. Preparation of HuNoV Nucleotide Sequence Dataset
2.5. Phylogenetic Tree Analyses
2.6. Stastiscal Analyses
2.7. Ethical Approval
3. Results
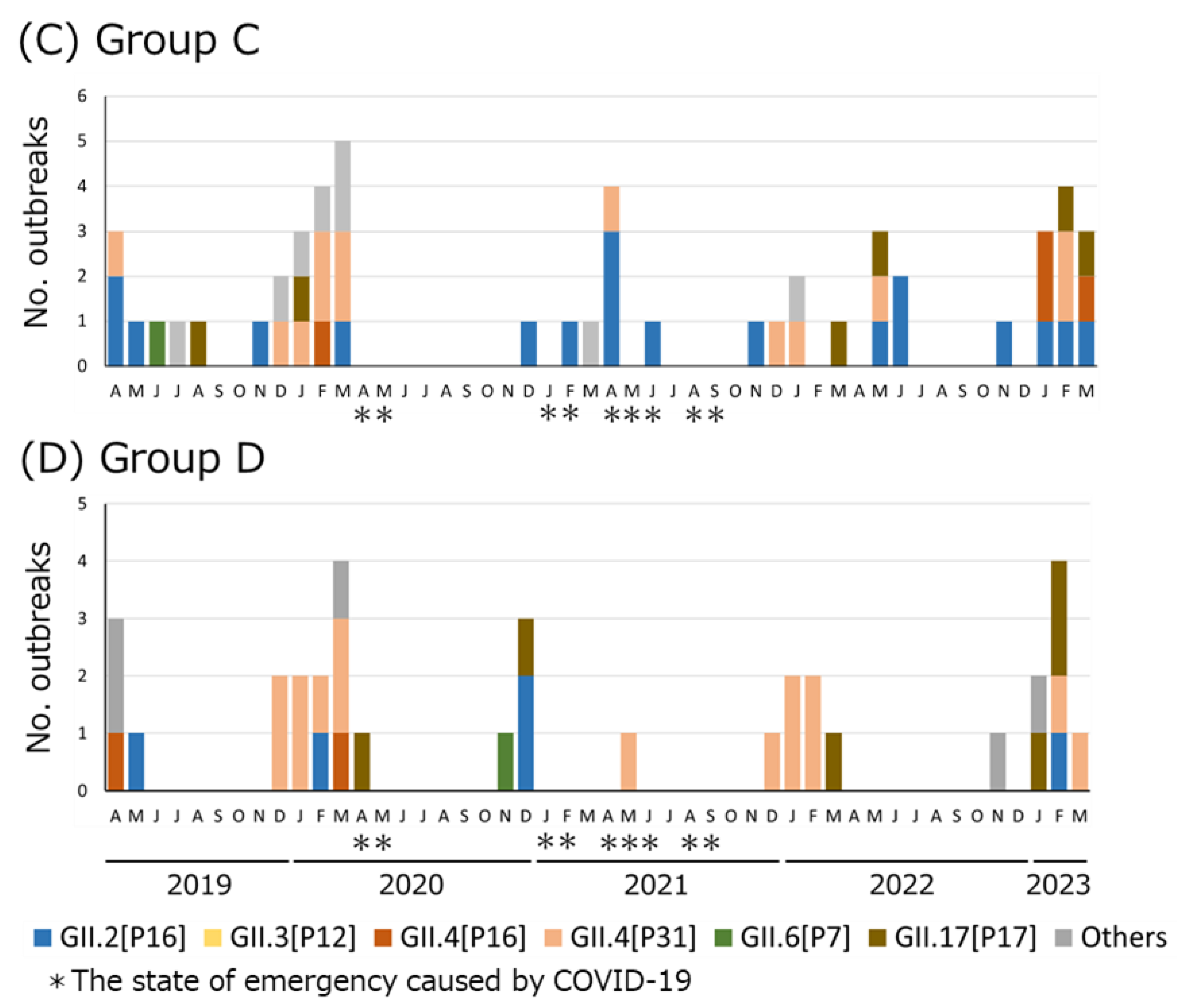
3.1. HuNoV Dual Typing in Each Group
3.2. Genotype/P-Type Combinations of HuNoV Detected in Osaka before and after COVID-19 Countermeasures
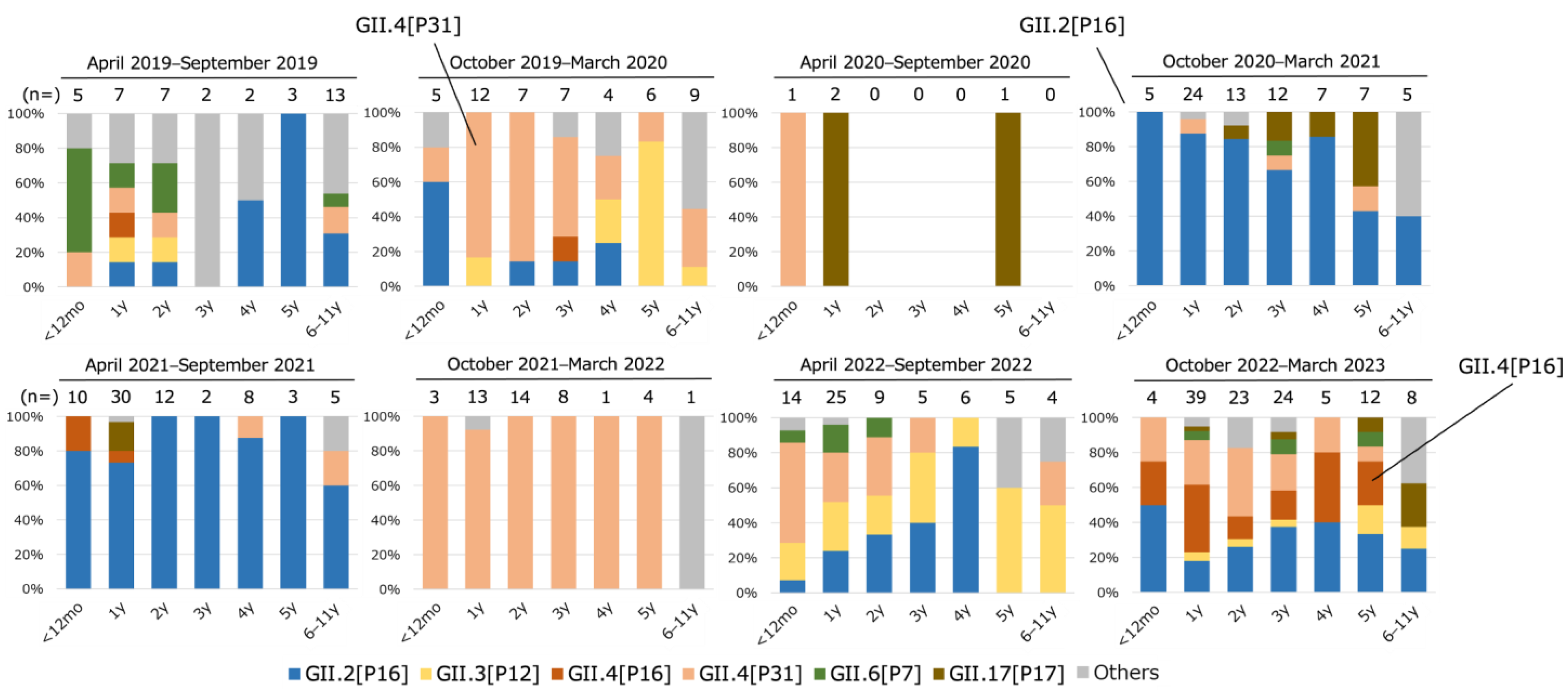
3.3. Distribution of Genotype/P-Type Combinations in Children at Each Age
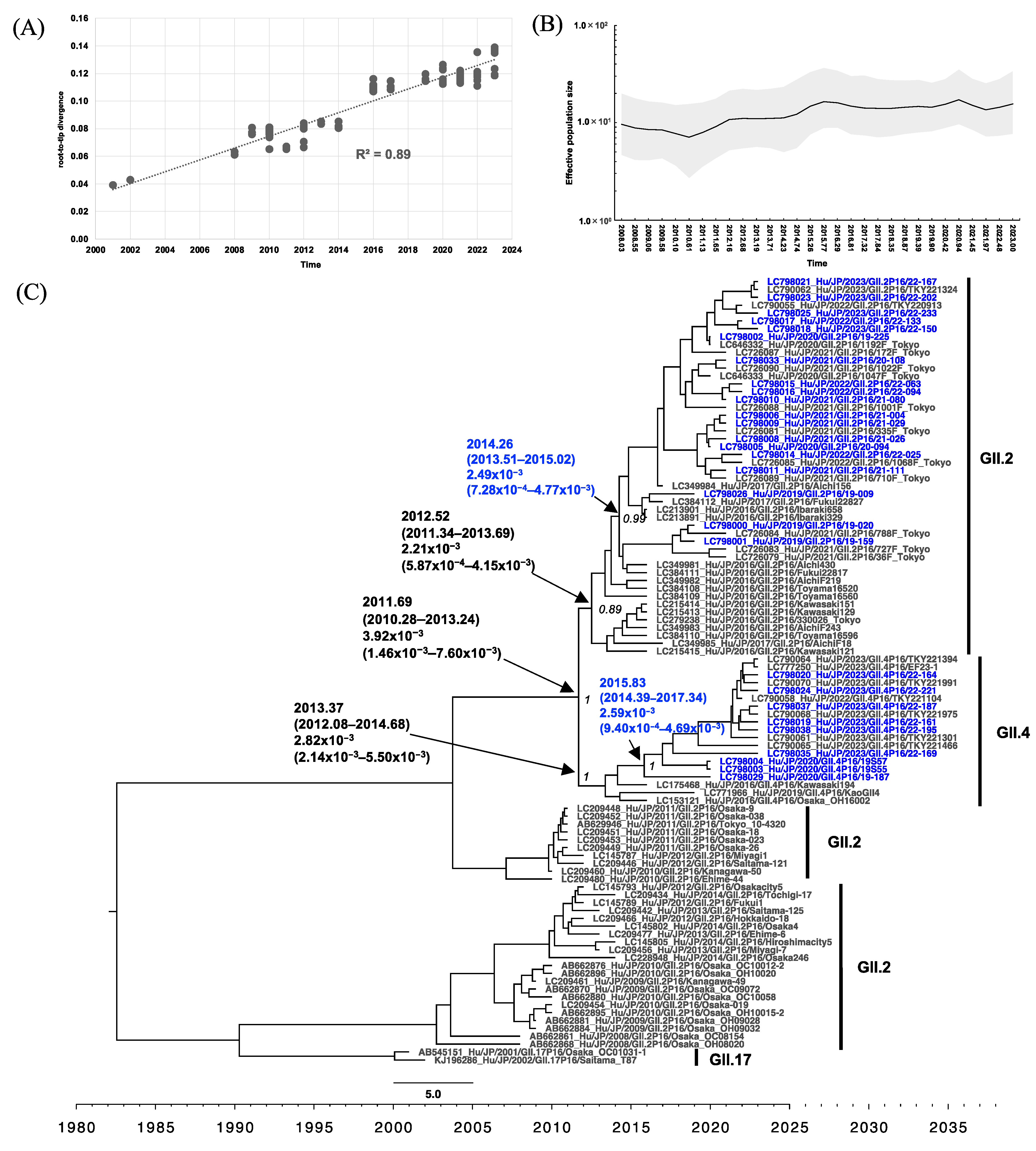
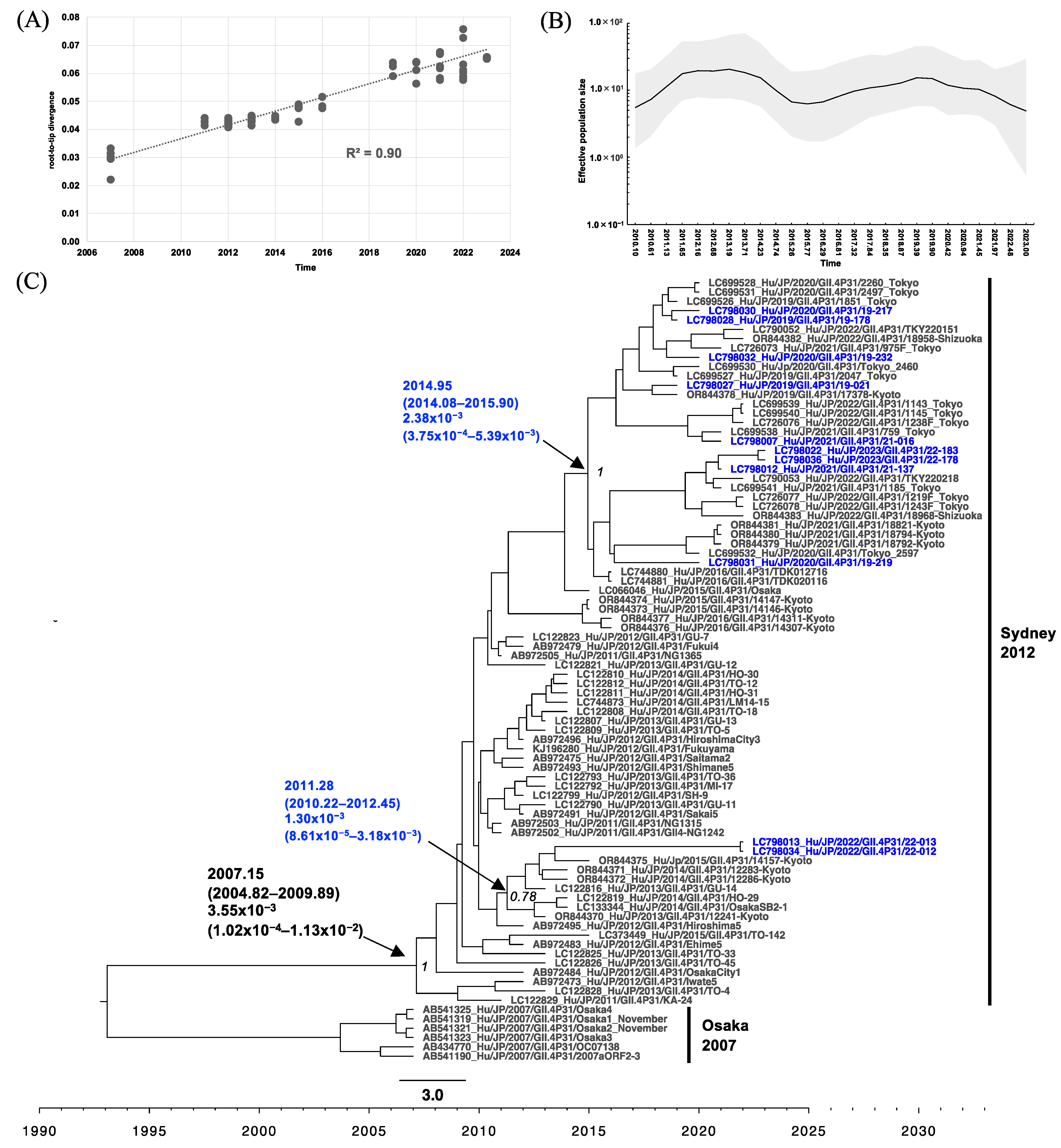
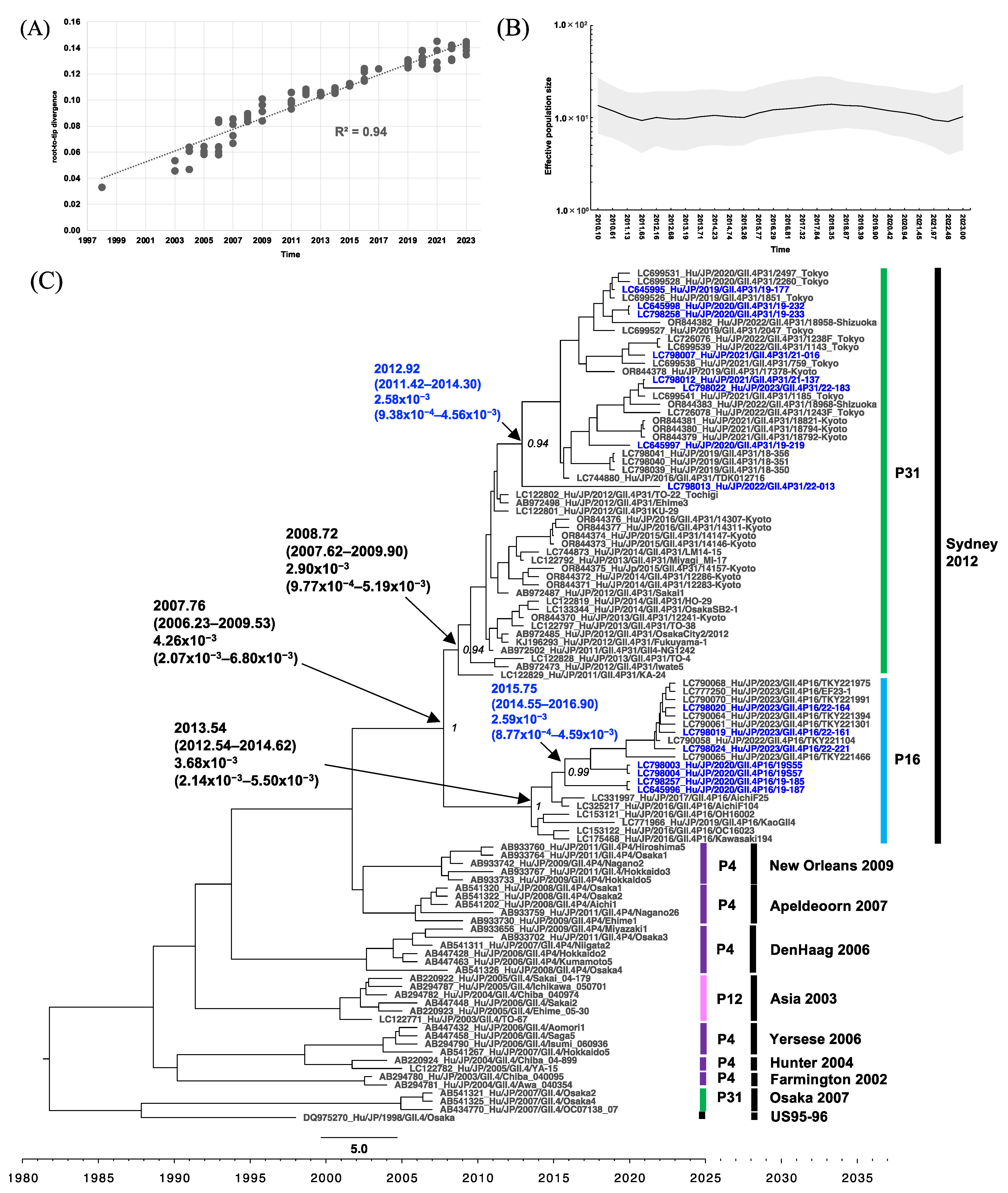
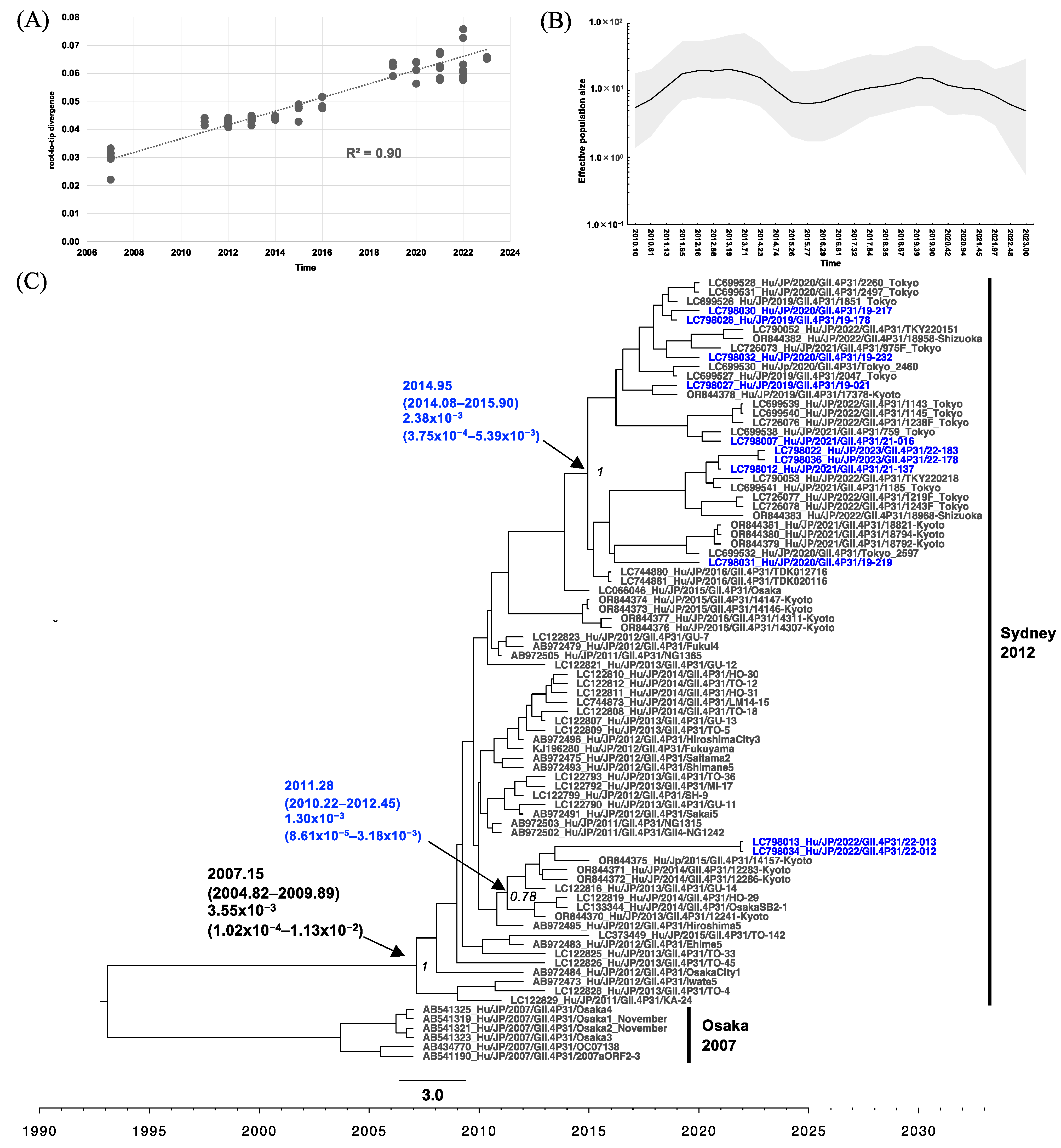
3.4. Phylogenetic Analysis of RdRp and VP1 of HuNoV
3.5. Analysis of RdRp Amino Acid Sequences and VP1 Antigenic Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, S.M.; Lopman, B.A.; Levy, K. A systematic review and meta-analysis of the global seasonality of norovirus. PLoS ONE 2013, 8, e75922. [Google Scholar] [CrossRef] [PubMed]
- Pires, S.M.; Fischer-Walker, C.L.; Lanata, C.F.; Devleesschauwer, B.; Hall, A.J.; Kirk, M.D.; Duarte, A.S.; Black, R.E.; Angulo, F.J. Aetiology-Specific Estimates of the Global and Regional Incidence and Mortality of Diarrhoeal Diseases Commonly Transmitted through Food. PLoS ONE 2015, 10, e0142927. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global Economic Burden of Norovirus Gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Hansman, G.S.; Clancy, L.E.; Tanaka, M.M.; Rawlinson, W.D.; White, P.A. Norovirus recombination in ORF1/ORF2 overlap. Emerg. Infect. Dis. 2005, 11, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Tanaka, M.M.; Boni, M.F.; Rawlinson, W.D.; White, P.A. Recombination within the pandemic norovirus GII.4 lineage. J. Virol. 2013, 87, 6270–6282. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; Jin, Z.; Chuang, Y.C.; Kao, C.C. Structure(s), function(s), and inhibition of the RNA-dependent RNA polymerase of noroviruses. Virus Res. 2017, 234, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Gao, Y.; Ford-Siltz, L.A.; Parra, G.I. Population Genomics of GII.4 Noroviruses Reveal Complex Diversification and New Antigenic Sites Involved in the Emergence of Pandemic Strains. mBio 2019, 10, e75922. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.; Ambert-Balay, K.; Botteldoorn, N.; Eden, J.S.; Fonager, J.; Hewitt, J.; Iritani, N.; Kroneman, A.; Vennema, H.; Vinjé, J.; et al. Indications for worldwide increased norovirus activity associated with emergence of a new variant of genotype II.4, late 2012. Euro Surveill. 2013, 18, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.J.; Noad, R.; Samuel, D.; Gray, J.J.; Roy, P.; Iturriza-Gómara, M. Characterisation of a GII-4 norovirus variant-specific surface-exposed site involved in antibody binding. Virol. J. 2009, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Van Loben Sels, J.M.; Green, K.Y. The Antigenic Topology of Norovirus as Defined by B and T Cell Epitope Mapping: Implications for Universal Vaccines and Therapeutics. Viruses 2019, 11, 432. [Google Scholar] [CrossRef] [PubMed]
- Iritani, N.; Kaida, A.; Abe, N.; Sekiguchi, J.; Kubo, H.; Takakura, K.; Goto, K.; Ogura, H.; Seto, Y. Increase of GII.2 norovirus infections during the 2009-2010 season in Osaka City, Japan. J. Med. Virol. 2012, 84, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Matsushima, Y.; Motoya, T.; Mizukoshi, F.; Ueki, Y.; Sakon, N.; Murakami, K.; Shimizu, T.; Okabe, N.; Nagata, N.; et al. Phylogeny and Immunoreactivity of Norovirus GII.P16-GII.2, Japan, Winter 2016-17. Emerg. Infect. Dis. 2018, 24, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Ao, Y.; Wang, J.; Ling, H.; He, Y.; Dong, X.; Wang, X.; Peng, J.; Zhang, H.; Jin, M.; Duan, Z. Norovirus GII.P16/GII.2-Associated Gastroenteritis, China, 2016. Emerg. Infect. Dis. 2017, 23, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Hasing, M.E.; Lee, B.E.; Qiu, Y.; Xia, M.; Pabbaraju, K.; Wong, A.; Tipples, G.; Jiang, X.; Pang, X.L. Changes in norovirus genotype diversity in gastroenteritis outbreaks in Alberta, Canada: 2012–2018. BMC Infect. Dis. 2019, 19, 177. [Google Scholar] [CrossRef] [PubMed]
- Niendorf, S.; Jacobsen, S.; Faber, M.; Eis-Hübinger, A.M.; Hofmann, J.; Zimmermann, O.; Höhne, M.; Bock, C.T. Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Euro Surveill. 2017, 22, 30447. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Phylogenetic Analyses Suggest that Factors Other Than the Capsid Protein Play a Role in the Epidemic Potential of GII.2 Norovirus. mSphere 2017, 2, e00187-17. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.L.; Zhu, Z.X.; Cui, J.L.; Yu, J.M. Evolutionary analyses of emerging GII.2[P16] and GII.4 Sydney [P16] noroviruses. Virus Evol. 2022, 8, veac030. [Google Scholar] [CrossRef] [PubMed]
- Ao, Y.; Cong, X.; Jin, M.; Sun, X.; Wei, X.; Wang, J.; Zhang, Q.; Song, J.; Yu, J.; Cui, J.; et al. Genetic Analysis of Reemerging GII.P16-GII.2 Noroviruses in 2016–2017 in China. J. Infect. Dis. 2018, 218, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.; de Graaf, M.; Al-Hello, H.; Allen, D.J.; Ambert-Balay, K.; Botteldoorn, N.; Brytting, M.; Buesa, J.; Cabrerizo, M.; Chan, M.; et al. Molecular surveillance of norovirus, 2005–2016: An epidemiological analysis of data collected from the NoroNet network. Lancet Infect. Dis. 2018, 18, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I. Emergence of norovirus strains: A tale of two genes. Virus Evol. 2019, 5, vez048. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.L.; Barclay, L.; Collins, N.R.; Wikswo, M.E.; Castro, C.J.; Magaña, L.C.; Gregoricus, N.; Marine, R.L.; Chhabra, P.; Vinjé, J. Genetic and Epidemiologic Trends of Norovirus Outbreaks in the United States from 2013 to 2016 Demonstrated Emergence of Novel GII.4 Recombinant Viruses. J. Clin. Microbiol. 2017, 55, 2208–2221. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, S.K.; de Andrade, J.; Miagostovich, M.P.; Fumian, T.M. Virological and Epidemiological Features of Norovirus Infections in Brazil, 2017–2018. Viruses 2021, 13, 1724. [Google Scholar] [CrossRef] [PubMed]
- Niendorf, S.; Faber, M.; Tröger, A.; Hackler, J.; Jacobsen, S. Diversity of Noroviruses throughout Outbreaks in Germany 2018. Viruses 2020, 12, 1157. [Google Scholar] [CrossRef] [PubMed]
- Anfruns-Estrada, E.; Sabaté, S.; Razquin, E.; Cornejo Sánchez, T.; Bartolomé, R.; Torner, N.; Izquierdo, C.; Soldevila, N.; Coronas, L.; Domínguez, À.; et al. Epidemiological and Genetic Characterization of Norovirus Outbreaks That Occurred in Catalonia, Spain, 2017–2019. Viruses 2022, 14, 488. [Google Scholar] [CrossRef] [PubMed]
- Lun, J.H.; Hewitt, J.; Yan, G.J.H.; Enosi Tuipulotu, D.; Rawlinson, W.D.; White, P.A. Recombinant GII.P16/GII.4 Sydney 2012 Was the Dominant Norovirus Identified in Australia and New Zealand in 2017. Viruses 2018, 10, 548. [Google Scholar] [CrossRef]
- Khamrin, P.; Kumthip, K.; Yodmeeklin, A.; Jampanil, N.; Phengma, P.; Yamsakul, P.; Okitsu, S.; Kobayashi, T.; Ushijima, H.; Maneekarn, N. Changing Predominance of Norovirus Recombinant Strains GII.2[P16] to GII.4[P16] and GII.4[P31] in Thailand, 2017 to 2018. Microbiol. Spectr. 2022, 10, e0044822. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.L.; Bonifacio, J.; Bucardo, F.; Buesa, J.; Bruggink, L.; Chan, M.C.; Fumian, T.M.; Giri, S.; Gonzalez, M.D.; Hewitt, J.; et al. Global Trends in Norovirus Genotype Distribution among Children with Acute Gastroenteritis. Emerg. Infect. Dis. 2021, 27, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Ao, Y.; Lu, L.; Xu, J. Emergence of GII.4 Sydney[P16]-like Norovirus-Associated Gastroenteritis, China, 2020–2022. Emerg. Infect. Dis. 2023, 29, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.T.K.; Khamrin, P.; Shimizu-Onda, Y.; Hoque, S.A.; Trinh, Q.D.; Komine-Aizawa, S.; Okitsu, S.; Maneekarn, N.; Hayakawa, S.; Yoshimune, K.; et al. Genetic diversity and declining norovirus prevalence in infants and children during Japan’s COVID-19 pandemic: A three-year molecular surveillance. Arch. Virol. 2023, 168, 231. [Google Scholar] [CrossRef] [PubMed]
- Sakon, N.; Takahashi, T.; Yoshida, T.; Shirai, T.; Komano, J. Impact of COVID-19 Countermeasures on Pediatric Infections. Microorganisms 2022, 10, 1947. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Ao, Y.; Jia, R.; Zhong, H.; Liu, P.; Xu, M.; Su, L.; Cao, L.; Xu, J. Changing predominance of norovirus strains in children with acute gastroenteritis in Shanghai, 2018–2021. Virol. Sin. 2023, 38, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Sakon, N.; Yamazaki, K.; Nakata, K.; Kanbayashi, D.; Yoda, T.; Mantani, M.; Kase, T.; Takahashi, K.; Komano, J. Impact of genotype-specific herd immunity on the circulatory dynamism of norovirus: A 10-year longitudinal study of viral acute gastroenteritis. J. Infect. Dis. 2015, 211, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, T.; Kojima, S.; Shinohara, M.; Uchida, K.; Fukushi, S.; Hoshino, F.B.; Takeda, N.; Katayama, K. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J. Clin. Microbiol. 2003, 41, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Obara, M.; Hasegawa, S.; Iwai, M.; Horimoto, E.; Nakamura, K.; Kurata, T.; Saito, N.; Oe, H.; Takizawa, T. Single base substitutions in the capsid region of the norovirus genome during viral shedding in cases of infection in areas where norovirus infection is endemic. J. Clin. Microbiol. 2008, 46, 3397–3403. [Google Scholar] [CrossRef]
- Fujii, M.; Yamamoto, J.; Mukai, H.; Fujita, M.; Tsukagoshi, H.; Yoshizumi, M.; Saito, M.; Kozawa, K.; Kimura, H. Detection and Quantitation of Norovirus Genome Using Real-Time RT-PCR. Jpn. J. Food Microbiol. 2011, 28, 139–142. (In Japanese) [Google Scholar] [CrossRef]
- Chhabra, P.; Browne, H.; Huynh, T.; Diez-Valcarce, M.; Barclay, L.; Kosek, M.N.; Ahmed, T.; Lopez, M.R.; Pan, C.Y.; Vinje, J. Single-step RT-PCR assay for dual genotyping of GI and GII norovirus strains. J. Clin. Virol. 2021, 134, 104689. [Google Scholar] [CrossRef] [PubMed]
- Motomura, K.; Oka, T.; Yokoyama, M.; Nakamura, H.; Mori, H.; Ode, H.; Hansman, G.S.; Katayama, K.; Kanda, T.; Tanaka, T.; et al. Identification of monomorphic and divergent haplotypes in the 2006–2007 norovirus GII/4 epidemic population by genomewide tracing of evolutionary history. J. Virol. 2008, 82, 11247–11262. [Google Scholar] [CrossRef] [PubMed]
- Shirai, T.; Sakon, N.; Takada, R.; Motomura, K. Epidemic and molecular epidemiological analysis of norovirus outbreaks in case of food poisoning in Osaka Prefecture (Fiscal 2019–2020 report). Ann. Rep. Osaka Inst. Pub Health 2021, 5, 18–24. (In Japanese) [Google Scholar]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.; Faber, M.; Altmann, B.; Mas Marques, A.; Bock, C.T.; Niendorf, S. Impact of the COVID-19 pandemic on norovirus circulation in Germany. Int. J. Med. Microbiol. 2024, 314, 151600. [Google Scholar] [CrossRef] [PubMed]
- Honjo, S.; Kuronuma, K.; Fujiya, Y.; Nakae, M.; Ukae, S.; Nihira, H.; Yamamoto, M.; Akane, Y.; Kondo, K.; Takahashi, S.; et al. Genotypes and transmission routes of noroviruses causing sporadic acute gastroenteritis among adults and children, Japan, 2015–2019. Infect. Genet. Evol. 2022, 104, 105348. [Google Scholar] [CrossRef] [PubMed]
- Kumazaki, M.; Usuku, S. Influence of herd immunity on norovirus: A long-term field study of repeated viral gastroenteritis outbreaks at the same facilities. BMC Infect. Dis. 2023, 23, 265. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Tohma, K.; Ford-Siltz, L.A.; Eguino, P.; Kendra, J.A.; Pilewski, K.A.; Gao, Y. Minimal Antigenic Evolution after a Decade of Norovirus GII.4 Sydney_2012 Circulation in Humans. J. Virol. 2023, 97, e0171622. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Ford-Siltz, L.A.; Kendra, J.A.; Parra, G.I. Dynamic immunodominance hierarchy of neutralizing antibody responses to evolving GII.4 noroviruses. Cell Rep. 2022, 39, 110689. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Matsushima, Y.; Nagasawa, K.; Motoya, T.; Ryo, A.; Kuroda, M.; Katayama, K.; Kimura, H. Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase Region in Norovirus Genogroup II. Front. Microbiol. 2018, 9, 3070. [Google Scholar] [CrossRef]
- Li, X.; Liu, H.; Rife Magalis, B.; Kosakovsky Pond, S.L.; Volz, E.M. Molecular Evolution of Human Norovirus GII.2 Clusters. Front. Microbiol. 2021, 12, 655567. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.K.C.; Kwok, K.; Zhang, L.Y.; Mohammad, K.N.; Lui, G.C.Y.; Lee, N.; Nelson, E.A.S.; Lai, R.W.M.; Leung, T.F.; Chan, P.K.S.; et al. Higher Viral Load of Emerging Norovirus GII.P16-GII.2 than Pandemic GII.4 and Epidemic GII.17, Hong Kong, China. Emerg. Infect. Dis. 2019, 25, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Qu, L.; Ettayebi, K.; Crawford, S.E.; Blutt, S.E.; Robertson, M.J.; Zeng, X.L.; Tenge, V.R.; Ayyar, B.V.; Karandikar, U.C.; et al. Human norovirus exhibits strain-specific sensitivity to host interferon pathways in human intestinal enteroids. Proc. Natl. Acad. Sci. USA 2020, 117, 23782–23793. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Infection Route | Specifications | No. of Outbreaks | No. of HuNoV Outbreaks (%) | No. of Samples | No. of Positives for HuNoV (%) |
|---|---|---|---|---|---|---|
| A | Sporadic | 0–14 y | - | - | 326 | 112 (34.4) |
| B | Human-to-human | Childcare facilities and schools | 258 | 182 (70.5) | 801 | 441 (55.1) |
| C | Suspected foodborne | - | 139 | 50 (36.0) | 690 | 185 (26.8) |
| D | Human-to-human | Nursing homes | 34 | 32 (94.1) | 105 | 89 (84.8) |
| Type | Name of Strain | Accession Number | Collection Year | Amino Acid Position | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 18 | 54 | 68 | 81 | 85 | 111 | 121 | 125 | 132 | 175 | 178 | 208 | 257 | 274 | 312 | 386 | 405 | 427 | 457 | 464 | 479 | 502 | |||||
| References | GII.2 [P16] | Santa Rosa 1764 | KY865306 | 2016 | L | G | K | K | S | A | A | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N |
| GII.4 [P16] | SH21-668 | OQ940080 | 2021 | L | G | R | K | S | A | A | H | A | N | I | K | A | A | I | T | N | K | T | R | T | E | N | |
| Samples in the Present Study | GII.2 [P16] | 19-009 | LC798026 | 2019 | L | G | K | K | S | A | A | H | V | N | I | K | A | A | T | A | N | K | N | K | T | E | N |
| 19-020 | LC798000 | 2019 | L | G | K | K | G | A | A | H | V | N | V | K | T | A | I | A | N | K | N | K | T | E | N | ||
| 19-159 | LC798001 | 2019 | L | G | K | K | G | A | A | H | V | N | V | K | A | A | I | A | D | K | N | K | T | E | N | ||
| 19-225 | LC798002 | 2020 | L | G | K | K | S | A | A | H | V | N | I | K | A | A | T | A | N | K | N | K | T | E | N | ||
| 20-094 | LC798005 | 2020 | L | G | K | K | S | A | A | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 20-108 | LC798033 | 2021 | L | G | K | K | S | A | A | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 21-004 | LC798006 | 2021 | I | G | K | K | S | A | A | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 21-026 | LC798008 | 2021 | L | D | K | R | S | A | A | Y | V | N | I | K | A | A | I | A | N | K | N | K | N | E | N | ||
| 21-029 | LC798009 | 2021 | L | G | K | K | S | A | A | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 21-080 | LC798010 | 2021 | L | G | K | K | S | A | V | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 21-111 | LC798011 | 2021 | L | G | K | K | S | A | A | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 22-025 | LC798014 | 2022 | L | G | K | K | S | A | A | H | V | N | I | K | A | T | I | A | N | K | N | K | T | E | N | ||
| 22-063 | LC798015 | 2022 | L | G | K | K | S | A | V | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 22-094 | LC798016 | 2022 | L | G | K | K | S | A | V | H | V | N | I | K | A | A | I | A | N | K | N | K | T | E | N | ||
| 22-150 | LC798018 | 2023 | L | G | K | K | S | S | A | Y | V | N | I | K | A | A | T | A | N | R | N | K | T | E | N | ||
| GII.4 [P16] | 19-187 | LC798029 | 2020 | L | S | K | K | S | A | A | H | V | N | I | R | A | A | I | A | N | K | S | R | T | E | S | |
| 19S55 | LC798003 | 2020 | L | G | R | K | S | A | A | H | A | N | I | K | A | A | I | T | N | K | N | R | T | E | N | ||
| 19S57 | LC798004 | 2020 | L | G | R | K | S | A | A | H | A | N | I | K | A | A | I | T | N | K | N | R | T | E | N | ||
| 22-161 | LC798019 | 2023 | L | G | R | K | S | A | A | H | V | S | I | K | T | A | I | A | D | K | N | R | T | G | N | ||
| 22-164 | LC798020 | 2023 | L | G | R | K | S | A | A | H | V | S | I | K | A | A | I | A | D | K | N | R | T | E | N | ||
| 22-221 | LC798024 | 2023 | L | G | R | K | S | A | A | H | V | S | I | K | A | A | I | A | D | K | N | R | T | E | N | ||
| Type | Name of Variant or Strain | Accession Number | Collection Year | Amino Acid Position | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Antigenic Site A | Antigenic Site C | Antigenic Site D | Antigenic Site E | Antigenic Site G | |||||||||||||||||||||||||||||||
| 294 | 295 | 296 | 297 | 298 | 368 | 372 | 373 | 339 | 340 | 341 | 375 | 376 | 377 | 378 | 393 | 394 | 395 | 396 | 397 | 407 | 411 | 412 | 413 | 414 | 352 | 355 | 356 | 357 | 359 | 364 | |||||
| References | GII.4[P31] | Sydney 2012_NSW0514 | JX459908 | 2012 | T | G | S | R | N | E | D | R | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R |
| GII.4[P16] | Sydney 2012_OH16002 | LC153121 | 2016 | T | G | S | R | N | E | D | H | R | T | D | F | E | A | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | |
| GII.4[P16] | SH21-668 | OQ940080 | 2021 | T | G | S | H | N | E | N | H | R | T | D | F | E | A | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | |
| Samples in the Present Study | GII.4[P31] | 19-177 | LC645995 | 2019 | T | G | S | H | N | E | N | N | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | P | Y | S | A | D | A | R |
| 19-219 | LC645997 | 2020 | T | G | S | H | N | E | N | N | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | P | Y | S | A | D | A | R | ||
| 19-232 * | LC645998 | 2020 | T | G | S | H | N | E | N | N | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | P | Y | S | A | D | A | R | ||
| 19-233 * | LC798258 | 2020 | T | G | S | H | N | E | N | N | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | P | Y | S | A | D | A | R | ||
| 21-016 | LC798007 | 2021 | T | G | S | H | N | E | N | N | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | P | Y | S | A | D | A | R | ||
| 21-137 | LC798012 | 2021 | T | G | S | H | N | E | N | N | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | P | Y | S | A | D | A | R | ||
| 22-013 | LC798013 | 2022 | T | G | S | H | N | E | N | H | R | T | D | F | E | A | N | G | T | T | H | R | S | R | S | T | P | Y | S | A | D | A | R | ||
| 22-183 | LC798022 | 2023 | T | G | S | H | N | E | N | N | R | T | D | F | E | A | N | G | T | T | H | R | S | R | N | T | P | Y | S | A | D | A | R | ||
| GII.4[P16] | 19-185 * | LC798257 | 2020 | T | G | S | R | N | E | D | H | R | T | D | F | E | V | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | |
| 19-187 * | LC645996 | 2020 | T | G | S | R | N | E | D | H | R | T | D | F | E | V | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | ||
| 19S55 | LC798003 | 2020 | T | G | S | H | N | E | N | H | R | T | D | F | E | A | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | ||
| 19S57 | LC798004 | 2020 | T | G | S | H | N | E | N | H | R | T | D | F | E | A | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | ||
| 22-161 | LC798019 | 2023 | T | G | S | H | N | E | N | H | R | T | D | F | E | A | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | ||
| 22-164 | LC798020 | 2023 | T | G | S | H | N | E | N | H | R | T | D | F | E | A | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | ||
| 22-221 | LC798024 | 2023 | T | G | S | H | N | E | N | H | R | T | D | F | E | A | N | S | T | T | H | R | S | R | N | T | H | Y | S | A | D | A | R | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirai, T.; Phadungsombat, J.; Ushikai, Y.; Yoshikaie, K.; Shioda, T.; Sakon, N. Epidemiological Features of Human Norovirus Genotypes before and after COVID-19 Countermeasures in Osaka, Japan. Viruses 2024, 16, 654. https://doi.org/10.3390/v16040654
Shirai T, Phadungsombat J, Ushikai Y, Yoshikaie K, Shioda T, Sakon N. Epidemiological Features of Human Norovirus Genotypes before and after COVID-19 Countermeasures in Osaka, Japan. Viruses. 2024; 16(4):654. https://doi.org/10.3390/v16040654
Chicago/Turabian StyleShirai, Tatsuya, Juthamas Phadungsombat, Yumi Ushikai, Kunihito Yoshikaie, Tatsuo Shioda, and Naomi Sakon. 2024. "Epidemiological Features of Human Norovirus Genotypes before and after COVID-19 Countermeasures in Osaka, Japan" Viruses 16, no. 4: 654. https://doi.org/10.3390/v16040654