
Unraveling the Properties of Phage Display Fab Libraries and Their Use in the Selection of Gliadin-Specific Probes by Applying High-Throughput Nanopore Sequencing
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Source Material: DNA Isolation and Purification
2.2. DNA Preparation for Sequencing Protocol
2.3. Primary Sequence Analysis
2.4. Secondary Analysis of Antibody Sequences
3. Results and Discussion
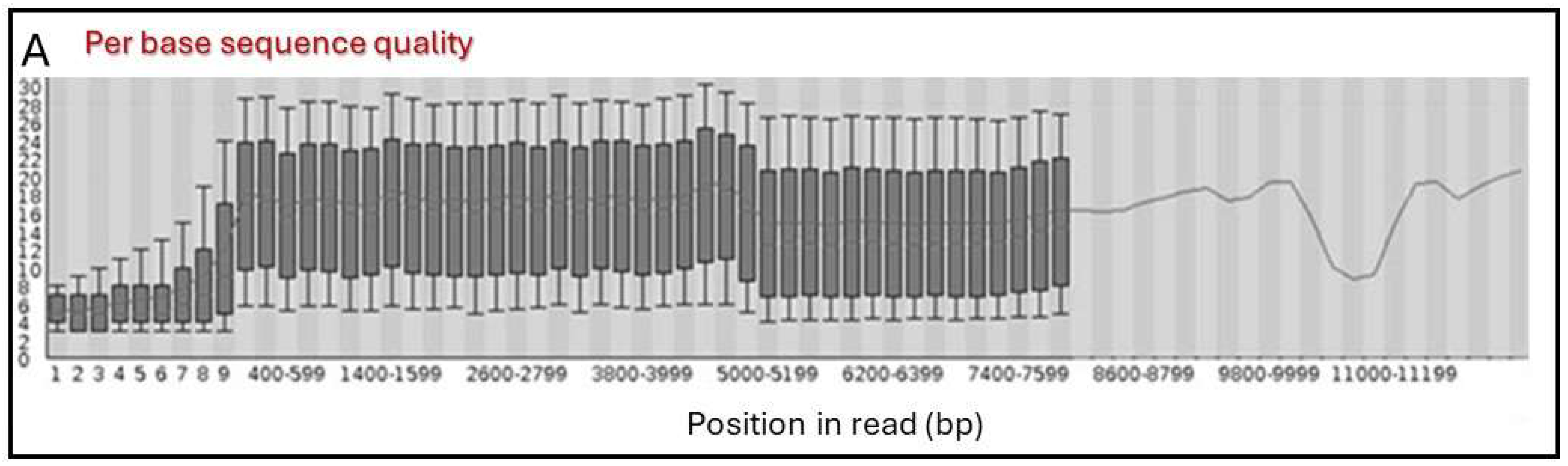
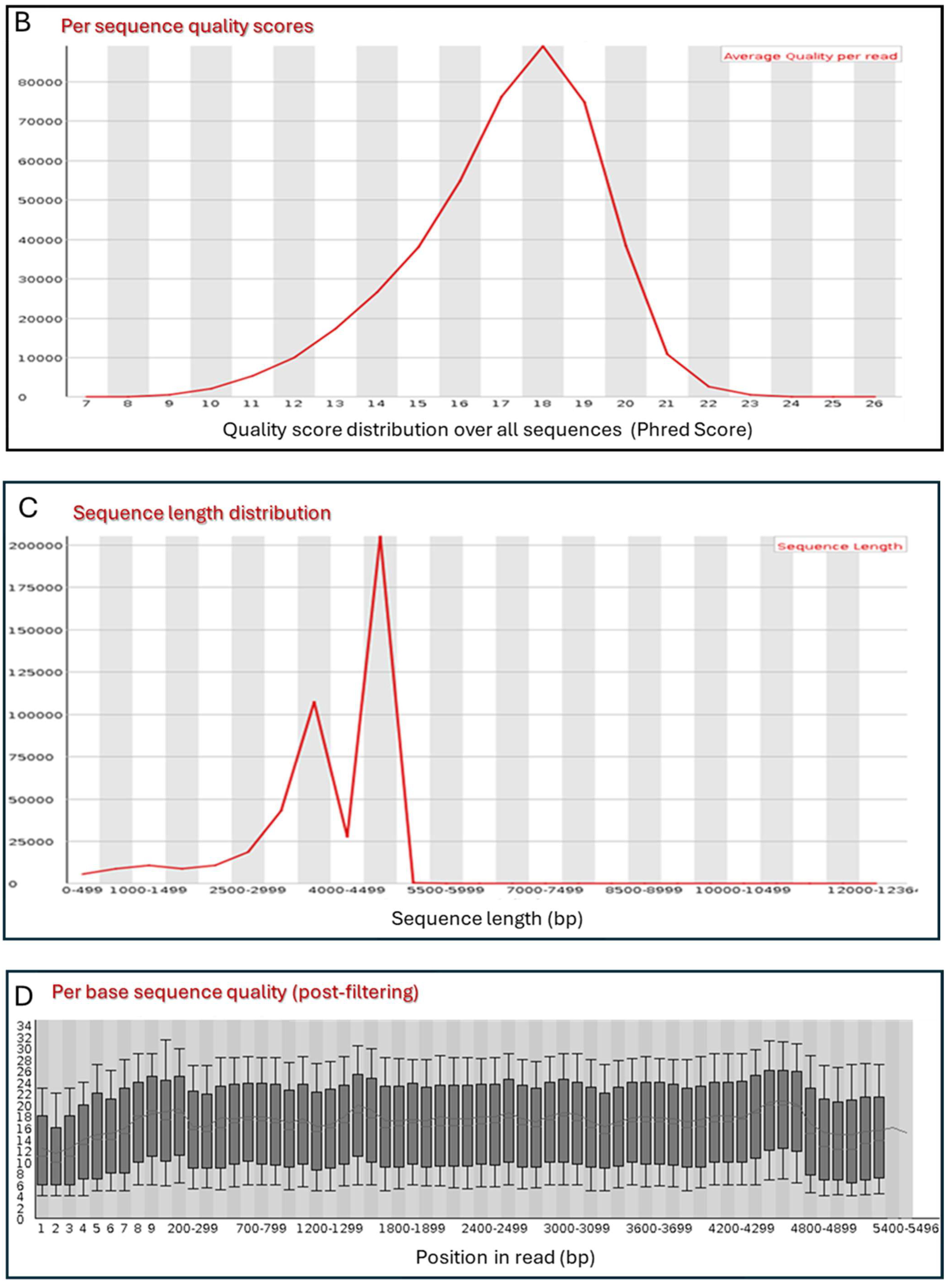
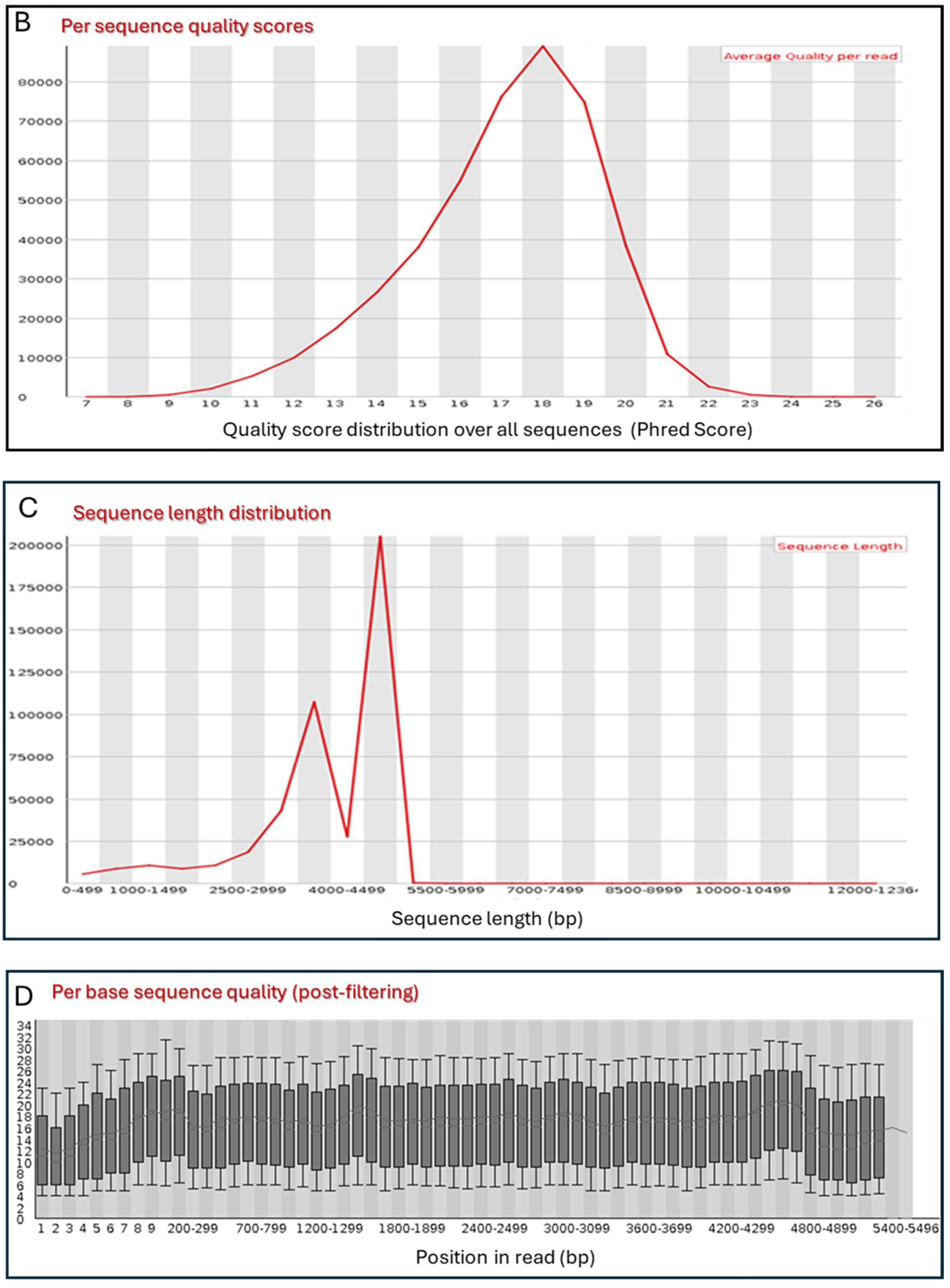
3.1. DNA Preparation
3.2. Primary Sequence Analysis
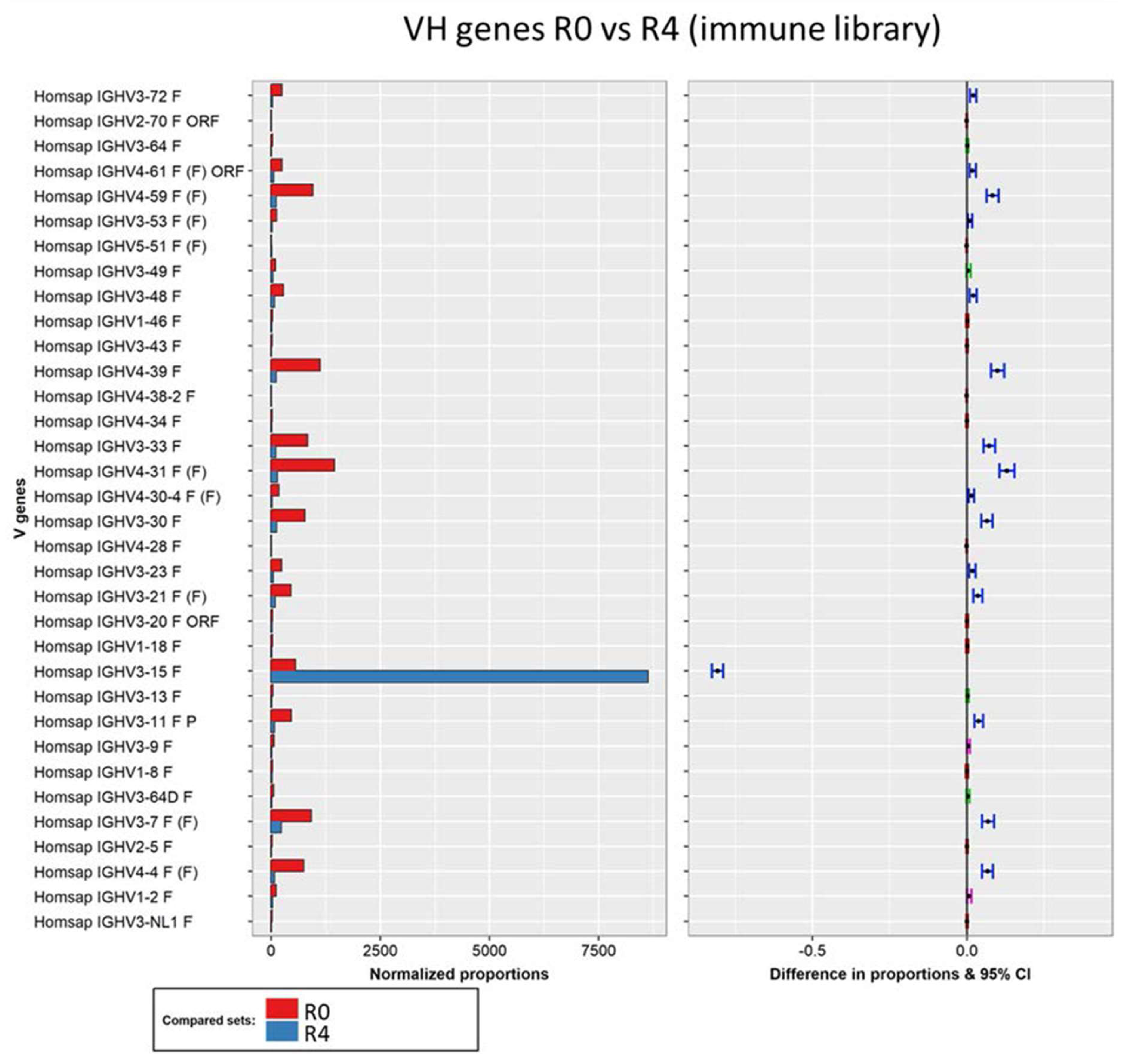
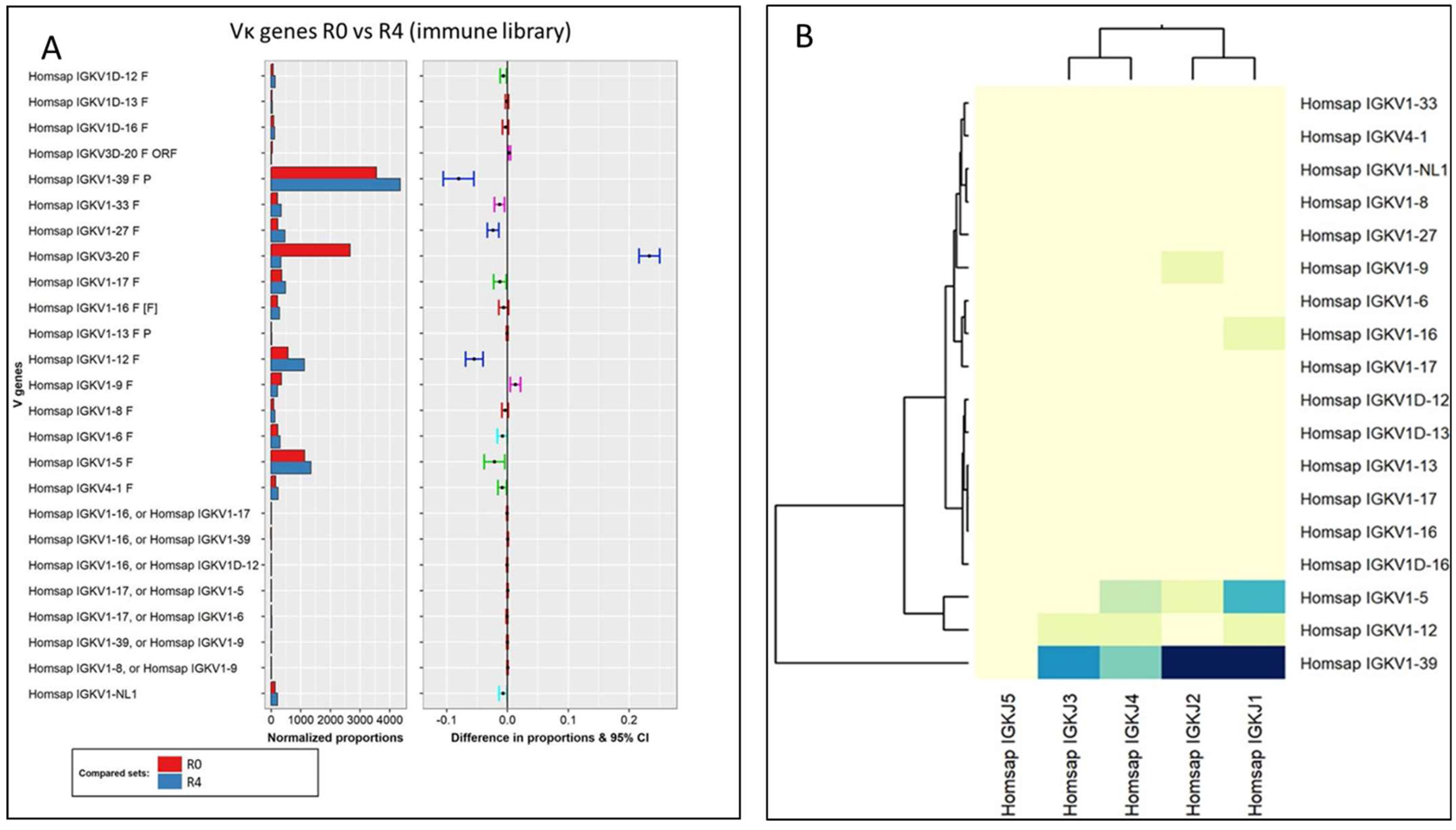
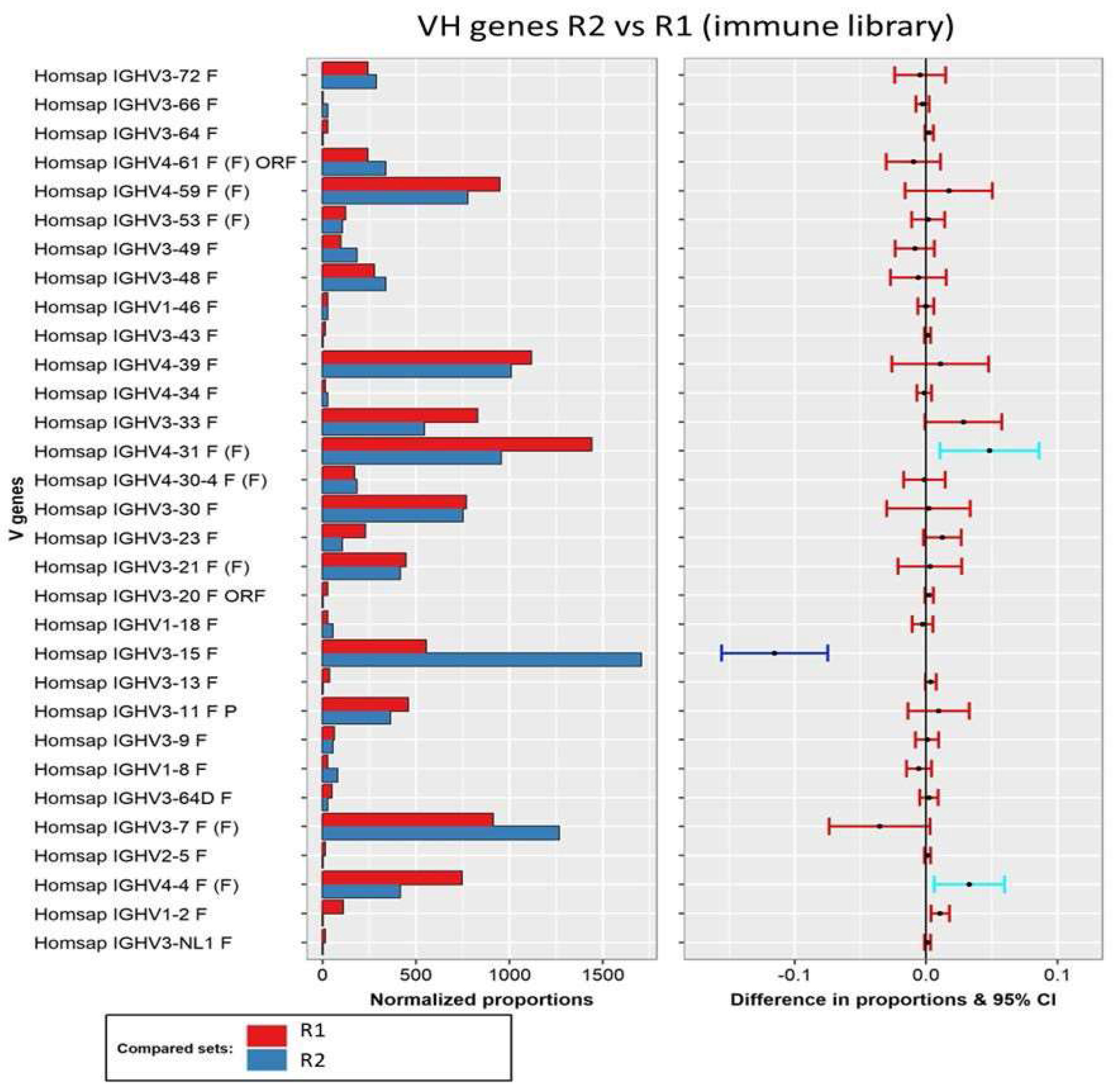
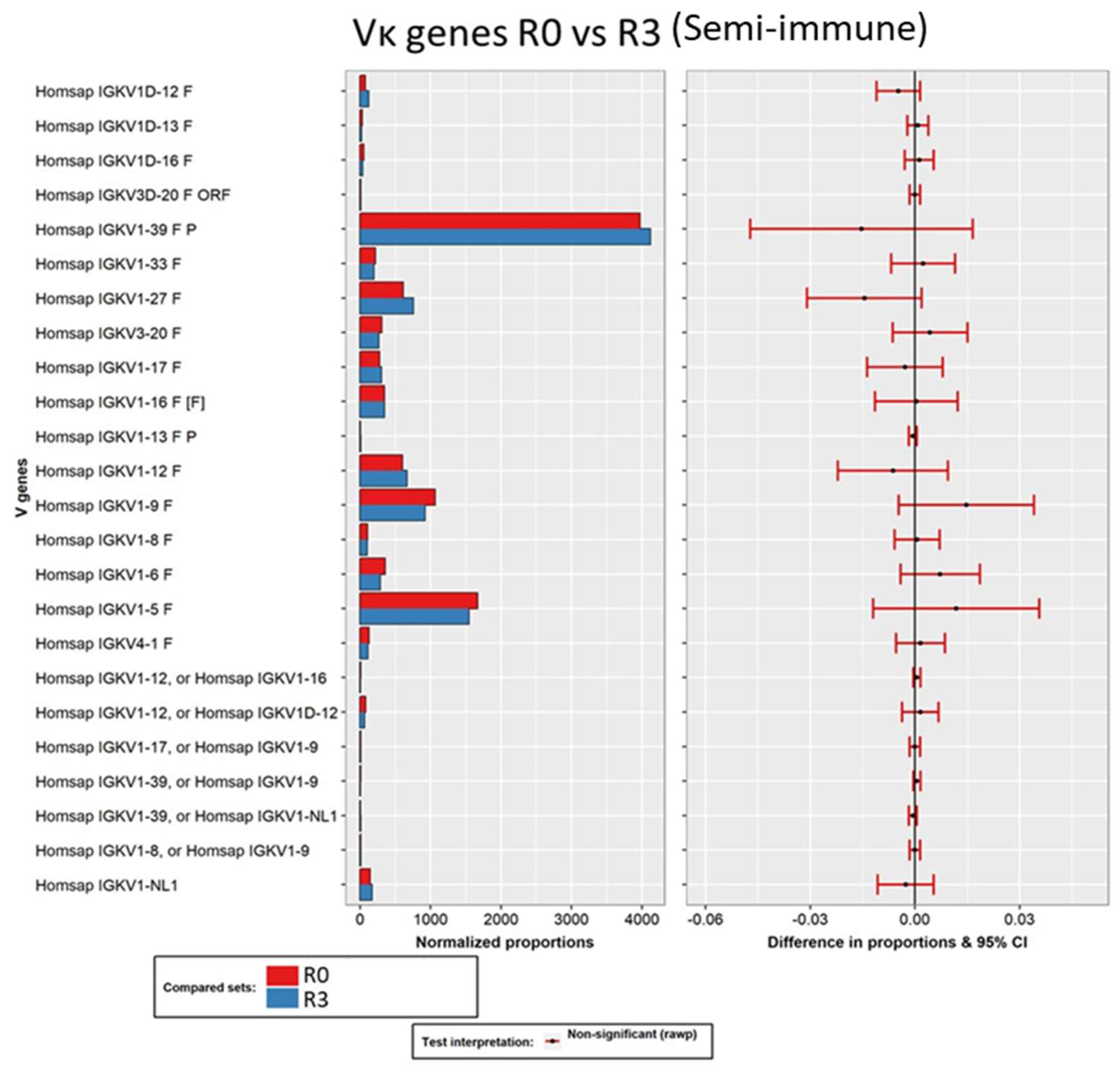
3.3. Secondary Analysis of Antibody Sequences and Biological Interpretation of the Results
3.3.1. Analysis of the Variable Domains
3.3.2. Analysis of the Constant Domains
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, L. Top companies and drugs by sales in 2022. Nat. Rev. Drug Discov. 2023, 22, 260. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef]
- Winter, G. Harnessing Evolution to Make Medicines (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14438–14445. [Google Scholar] [CrossRef]
- Smith, G.P. Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14428–14437. [Google Scholar] [CrossRef] [PubMed]
- Glanville, J.; D’Angelo, S.; Khan, T.A.; Reddy, S.T.; Naranjo, L.; Ferrara, F.; Bradbury, A.R.M. Deep sequencing in library selection projects: What insight does it bring? Curr. Opin. Struct. Biol. 2015, 33, 146–160. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.; Kumar, S.; Naranjo, L.; Ferrara, F.; Kiss, C.; Bradbury, A.R. From deep sequencing to actual clones. Protein Eng. Des. Sel. 2014, 27, 301–307. [Google Scholar] [CrossRef]
- Barreto, K.; Maruthachalam, B.V.; Hill, W.; Hogan, D.; Sutherland, A.R.; Kusalik, A.; Fonge, H.; DeCoteau, J.F.; Geyer, C.R. Next-generation sequencing-guided identification and reconstruction of antibody CDR combinations from phage selection outputs. Nucleic Acids Res. 2019, 47, e50. [Google Scholar] [CrossRef] [PubMed]
- Matochko, W.L.; Derda, R. Next-generation sequencing of phage-displayed peptide libraries. Methods Mol. Biol. 2015, 1248, 249–266. [Google Scholar] [CrossRef]
- Chen, P.; Sun, Z.; Wang, J.; Liu, X.; Bai, Y.; Chen, J.; Liu, A.; Qiao, F.; Chen, Y.; Yuan, C.; et al. Portable nanopore-sequencing technology: Trends in development and applications. Front. Microbiol. 2023, 14, 1043967. [Google Scholar] [CrossRef]
- Joyce, C.; Burton, D.R.; Briney, B. Comparisons of the antibody repertoires of a humanized rodent and humans by high throughput sequencing. Sci. Rep. 2020, 10, 1120. [Google Scholar] [CrossRef]
- Roth, D.B. V(D)J Recombination: Mechanism, Errors, and Fidelity. Microbiol. Spectr. 2014, 2, 313–324. [Google Scholar] [CrossRef]
- Hwang, J.K.; Alt, F.W.; Yeap, L.S. Related Mechanisms of Antibody Somatic Hypermutation and Class Switch Recombination. Microbiol. Spectr. 2015, 3, 325–348. [Google Scholar] [CrossRef]
- Barbas, C.F. Phage Display: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.; De Giorgio, R.; Catassi, C.; Fasano, A. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 142. [Google Scholar] [CrossRef]
- Sapone, A.; Bai, J.C.; Ciacci, C.; Dolinsek, J.; Green, P.H.; Hadjivassiliou, M.; Kaukinen, K.; Rostami, K.; Sanders, D.S.; Schumann, M.; et al. Spectrum of gluten-related disorders: Consensus on new nomenclature and classification. BMC Med. 2012, 10, 13. [Google Scholar] [CrossRef]
- Itzlinger, A.; Branchi, F.; Elli, L.; Schumann, M. Gluten-free diet in celiac disease—Forever and for all? Nutrients 2018, 10, 1796. [Google Scholar] [CrossRef]
- Garcia-Calvo, E.; García-García, A.; Rodríguez-Gómez, S.; Farrais, S.; Martín, R.; García, T. Development of a new recombinant antibody, selected by phage-display technology from a celiac patient library, for detection of gluten in foods. Curr. Res. Food Sci. 2023, 7, 100578. [Google Scholar] [CrossRef]
- Garcia-Calvo, E.; García-García, A.; Rodríguez, S.; Farrais, S.; Martín, R.; García, T. Construction of a Fab library merging chains from semisynthetic and immune origin, suitable for developing new tools for gluten immunodetection in food. Foods 2022, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Brochet, X.; Lefranc, M.P.; Giudicelli, V. IMGT/V-QUEST: The highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008, 36, W503–W508. [Google Scholar] [CrossRef]
- Lefranc, M.-P.; Lefranc, G. Immunoglobulins or Antibodies: IMGT® Bridging Genes, Structures and Functions. Biomedicines 2020, 8, 319. [Google Scholar] [CrossRef]
- Aouinti, S.; Malouche, D.; Giudicelli, V.; Kossida, S.; Lefranc, M.-P. IMGT/HighV-QUEST Statistical Significance of IMGT Clonotype (AA) Diversity per Gene for Standardized Comparisons of Next Generation Sequencing Immunoprofiles of Immunoglobulins and T Cell Receptors. PLoS ONE 2015, 10, e0142353. [Google Scholar] [CrossRef] [PubMed]
- Aouinti, S.; Giudicelli, V.; Duroux, P.; Malouche, D.; Kossida, S.; Lefranc, M.P. IMGT/StatClonotype for pairwise evaluation and visualization of NGS IG and TR IMGT Clonotype (AA) diversity or expression from IMGT/HighV-QUEST. Front. Immunol. 2016, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Lowden, M.J.; Henry, K.A. Oxford nanopore sequencing enables rapid discovery of single-domain antibodies from phage display libraries. BioTechniques 2018, 65, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Garaev, M.M.; Bobkov, A.F.; Bobkova, A.F.; Kalinin, V.N.; Smirnov, V.D.; Khudakov, Y.E.; Tikchonenko, T.I. The site-specific deletion in plasmid pBR322. Gene 1982, 18, 21–28. [Google Scholar] [CrossRef]
- Bottaro, G.; Volta, U.; Spina, M.; Rotolo, N.; Sciacca, A.; Musumeci, S. Antibody Pattern in Childhood Celiac Disease. J. Pediatr. Gastroenterol. Nutr. 1997, 24, 559–562. [Google Scholar] [PubMed]
- D’Angelo, S.; Mignone, F.; Deantonio, C.; Di Niro, R.; Bordoni, R.; Marzari, R.; De Bellis, G.; Not, T.; Ferrara, F.; Bradbury, A.; et al. Profiling celiac disease antibody repertoire. Clin. Immunol. 2013, 148, 99–109. [Google Scholar] [CrossRef]
- Erasmus, M.F.; D’Angelo, S.; Ferrara, F.; Naranjo, L.; Teixeira, A.A.; Buonpane, R.; Stewart, S.M.; Nastri, H.G.; Bradbury, A.R.M. A single donor is sufficient to produce a highly functional in vitro antibody library. Commun. Biol. 2021, 4, 350. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Calvo, E.; García-García, A.; Rodríguez, S.; Takkinen, K.; Martín, R.; García, T. Production and Characterization of Novel Fabs Generated from Different Phage Display Libraries as Probes for Immunoassays for Gluten Detection in Food. Foods 2023, 12, 3274. [Google Scholar] [CrossRef]
- Shi, B.; Dong, X.; Ma, Q.; Sun, S.; Ma, L.; Yu, J.; Wang, X.; Pan, J.; He, X.; Su, D.; et al. The usage of human IGHJ genes follows a particular non-random selection: The recombination signal sequence may affect the usage of human IGHJ genes. Front. Genet. 2020, 11, 524413. [Google Scholar] [CrossRef]
- Kim, S.; Park, I.; Park, S.G.; Cho, S.; Kim, J.H.; Ipper, N.S.; Choi, S.S.; Lee, E.S.; Hong, H.J. Generation, diversity determination, and application to antibody selection of a human naïve Fab library. Mol. Cells 2017, 40, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Steinsbø, Ø.; Dunand, C.J.H.; Huang, M.; Mesin, L.; Salgado-Ferrer, M.; Lundin, K.E.A.; Jahnsen, J.; Wilson, P.C.; Sollid, L.M. Restricted VH/VL usage and limited mutations in gluten-specific IgA of coeliac disease lesion plasma cells. Nat. Commun. 2014, 5, 4041. [Google Scholar] [CrossRef]
- Maclennan, I.C.M.; Toellner, K.M.; Cunningham, A.F.; Serre, K.; Sze, D.M.Y.; Zúñiga, E.; Cook, M.C.; Vinuesa, C.G. Extrafollicular antibody responses. Immunol. Rev. 2003, 194, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Dutta, P.R.; Cerasoli, D.M.; Kelsoe, G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J. Exp. Med. 1998, 187, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Ho, F.; Lortan, J.E.; MacLennan, I.C.; Khan, M. Distinct short-lived and long-lived antibody-producing cell populations. Eur. J. Immunol. 1986, 16, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Rigby, R.J.; Zotos, D.; Tsai, L.M.; Kawamoto, S.; Marshall, J.L.; Ramiscal, R.R.; Chan, T.D.; Gatto, D.; Brink, R.; et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J. Exp. Med. 2011, 208, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Loh, R.K.; Vale, S.; McLean-Tooke, A. Quantitative serum immunoglobulin tests. Aust. Fam. Physician 2013, 42, 195–198. [Google Scholar]
- Volta, U.; Molinaro, N.; Fratangelo, D.; Bianchi, F.B. IgA subclass antibodies to gliadin in serum and intestinal juice of patients with coeliac disease. Clin. Exp. Immunol. 2008, 80, 192–195. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Germline | Library | % of Reads with the Mismatch | Amino Acid Changes |
|---|---|---|---|---|
| 13 | G | C | 92 | V → L |
| 15 | G | C | 91 | V → A |
| 92 | A | G | 91 | N → T |
| 99 | G | T | 93 | W → C |
| 150 | T | G | 96 | R → R |
| 176 | C | A | 91 | T → F |
| 188 | C | G | 93 | A → G |
| 193 | C | T | 95 | P → S |
| 252 | A | G | 96 | Q → Q |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Calvo, E.; García-García, A.; Rodríguez, S.; Martín, R.; García, T. Unraveling the Properties of Phage Display Fab Libraries and Their Use in the Selection of Gliadin-Specific Probes by Applying High-Throughput Nanopore Sequencing. Viruses 2024, 16, 686. https://doi.org/10.3390/v16050686
Garcia-Calvo E, García-García A, Rodríguez S, Martín R, García T. Unraveling the Properties of Phage Display Fab Libraries and Their Use in the Selection of Gliadin-Specific Probes by Applying High-Throughput Nanopore Sequencing. Viruses. 2024; 16(5):686. https://doi.org/10.3390/v16050686
Chicago/Turabian StyleGarcia-Calvo, Eduardo, Aina García-García, Santiago Rodríguez, Rosario Martín, and Teresa García. 2024. "Unraveling the Properties of Phage Display Fab Libraries and Their Use in the Selection of Gliadin-Specific Probes by Applying High-Throughput Nanopore Sequencing" Viruses 16, no. 5: 686. https://doi.org/10.3390/v16050686