CCR5: From Natural Resistance to a New Anti-HIV Strategy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
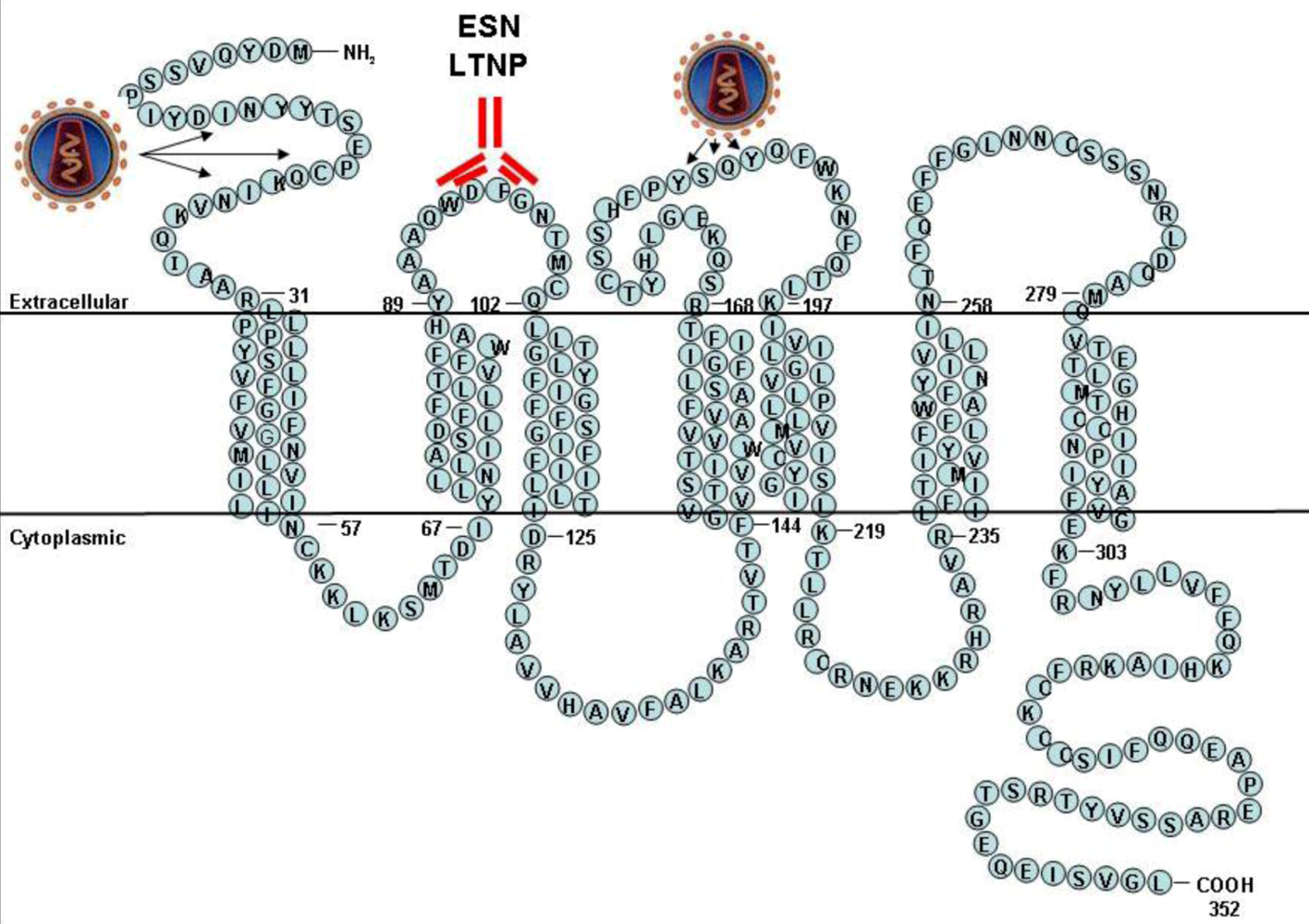
2. CCR5 functions

3. CCR5 deletion and its consequences
4. CCR5 role in HIV infection
4.1. CCR5 vs. CXCR4
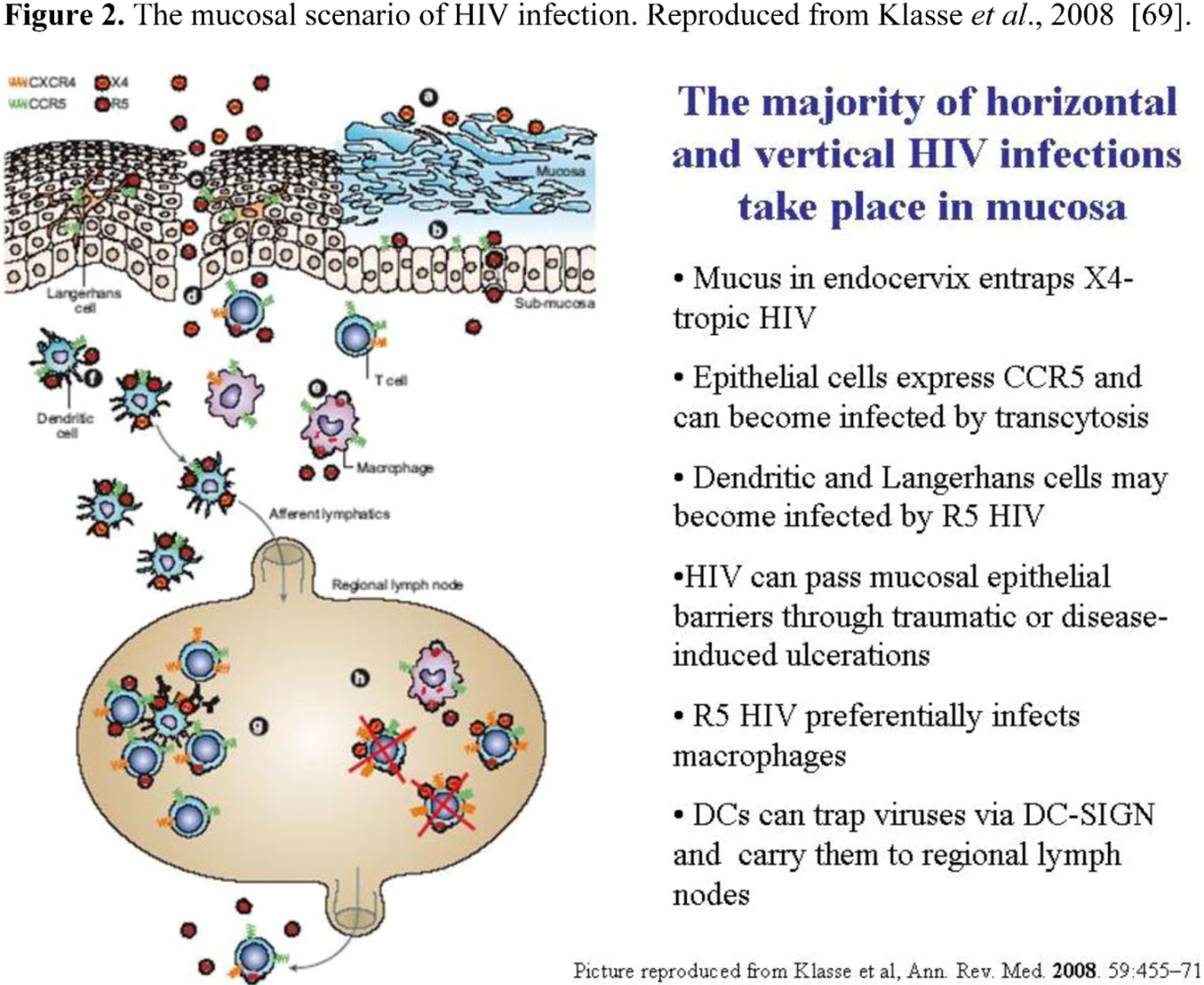
4.2. CCR5 in mucosal HIV transmission

5. Natural history of individuals carrying anti-CCR5 antibodies

6. Strategies for CCR5 targeting
6.1. Small molecule inhibitors

6.2. Monoclonal antibodies to CCR5
6.3. Engineered chemokines
6.4. Anti-CCR5 vaccination
7. Conclusion and perspectives for a vaccination intervention
Acknowledgments
References
- Lodowski, D.T.; Palczewski, K. Chemokine receptors and other G protein-coupled receptors. Curr. Opin. HIV AIDS 2009, 4, 88–95. [Google Scholar] [CrossRef]
- Corbeau, P.; Reynes, J. CCR5 antagonism in HIV infection: ways, effects, and side effects. Aids 2009, 23, 1931–1943. [Google Scholar] [CrossRef] [PubMed]
- Trifilo, M.J.; Bergmann, C.C.; Kuziel, W.A.; Lane, T.E. CC chemokine ligand 3 (CCL3) regulates CD8(+)-T-cell effector function and migration following viral infection. J. Virol. 2003, 77, 4004–4014. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G. The biology of CCR5 and CXCR4. Curr. Opin. HIV AIDS 2009, 4, 96–103. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kulkarni, H.; Bolivar, H.; Mangano, A.; Sanchez, R.; Catano, G.; Nibbs, R.J.; Freedman, B.I.; Quinones, M.P.; Bamshad, M.J.; Murthy, K.K.; Rovin, B.H.; Bradley, W.; Clark, R.A.; Anderson, S.A.; O'Connell R, J.; Agan, B.K.; Ahuja, S.S.; Bologna, R.; Sen, L.; Dolan, M.J.; Ahuja, S.K. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005, 307, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Contento, R.L.; Molon, B.; Boularan, C.; Pozzan, T.; Manes, S.; Marullo, S.; Viola, A. CXCR4-CCR5: a couple modulating T cell functions. Proc. Natl. Acad. Sci. USA 2008, 105, 10101–10106. [Google Scholar] [CrossRef]
- Bacon, K.B.; Premack, B.A.; Gardner, P.; Schall, T.J. Activation of dual T cell signaling pathways by the chemokine RANTES. Science 1995, 269, 1727–1730. [Google Scholar] [PubMed]
- Castellino, F.; Huang, A.Y.; Altan-Bonnet, G.; Stoll, S.; Scheinecker, C.; Germain, R.N. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 2006, 440, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Taub, D.D.; Ortaldo, J.R.; Turcovski-Corrales, S.M.; Key, M.L.; Longo, D.L.; Murphy, W.J. Beta chemokines costimulate lymphocyte cytolysis, proliferation, and lymphokine production. J. Leukoc. Biol. 1996, 59, 81–89. [Google Scholar] [PubMed]
- Huttenrauch, F.; Pollok-Kopp, B.; Oppermann, M. G protein-coupled receptor kinases promote phosphorylation and beta-arrestin-mediated internalization of CCR5 homo- and hetero-oligomers. J. Biol. Chem. 2005, 280, 37503–37515. [Google Scholar] [CrossRef] [PubMed]
- Shideman, C.R.; Hu, S.; Peterson, P.K.; Thayer, S.A. CCL5 evokes calcium signals in microglia through a kinase-, phosphoinositide-, and nucleotide-dependent mechanism. J. Neurosci. Res. 2006, 83, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Signoret, N.; Hewlett, L.; Wavre, S.; Pelchen-Matthews, A.; Oppermann, M.; Marsh, M. Agonist-induced endocytosis of CC chemokine receptor 5 is clathrin dependent. Mol. Biol. Cell 2005, 16, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.; Kelly, E.; Strange, P.G. Pathways for internalization and recycling of the chemokine receptor CCR5. Blood 2002, 99, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Manes, S.; del Real, G.; Lacalle, R.A.; Lucas, P.; Gomez-Mouton, C.; Sanchez-Palomino, S.; Delgado, R.; Alcami, J.; Mira, E.; Martinez, A.C. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000, 1, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Signoret, N.; Pelchen-Matthews, A.; Mack, M.; Proudfoot, A.E.; Marsh, M. Endocytosis and recycling of the HIV coreceptor CCR5. J. Cell Biol. 2000, 151, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Mettling, C.; Portales, P.; Rouzier, R.; Clot, J.; Reynes, J.; Corbeau, P. The chemokine CCL5 regulates the in vivo cell surface expression of its receptor, CCR5. Aids 2008, 22, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Rose, J.J.; Lodge, R.; Murphy, P.M.; Foley, J.F. Distinct mechanisms of agonist-induced endocytosis for human chemokine receptors CCR5 and CXCR4. Mol. Biol. Cell 2003, 14, 3305–3324. [Google Scholar] [CrossRef] [PubMed]
- Steffens, C.M.; Hope, T.J. Localization of CD4 and CCR5 in living cells. J. Virol. 2003, 77, 4985–4991. [Google Scholar] [CrossRef] [PubMed]
- Trkola, A.; Dragic, T.; Arthos, J.; Binley, J.M.; Olson, W.C.; Allaway, G.P.; Cheng-Mayer, C.; Robinson, J.; Maddon, P.J.; Moore, J.P. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 1996, 384, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Gerard, N.P.; Wyatt, R.; Choe, H.; Parolin, C.; Ruffing, N.; Borsetti, A.; Cardoso, A.A.; Desjardin, E.; Newman, W.; Gerard, C.; Sodroski, J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 1996, 384, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Achour, L.; Scott, M.G.; Shirvani, H.; Thuret, A.; Bismuth, G.; Labbe-Jullie, C.; Marullo, S. CD4-CCR5 interaction in intracellular compartments contributes to receptor expression at the cell surface. Blood 2009, 113, 1938–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issafras, H.; Angers, S.; Bulenger, S.; Blanpain, C.; Parmentier, M.; Labbe-Jullie, C.; Bouvier, M.; Marullo, S. Constitutive agonist-independent CCR5 oligomerization and antibody-mediated clustering occurring at physiological levels of receptors. J. Biol. Chem. 2002, 277, 34666–34673. [Google Scholar] [CrossRef] [PubMed]
- Benkirane, M.; Jin, D.Y.; Chun, R.F.; Koup, R.A.; Jeang, K.T. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J. Biol. Chem. 1997, 272, 30603–30606. [Google Scholar] [CrossRef] [PubMed]
- Mellado, M.; Rodriguez-Frade, J.M.; Vila-Coro, A.J.; de Ana, A.M.; Martinez, A.C. Chemokine control of HIV-1 infection. Nature 1999, 400, 723–724. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Chuang, L.F.; Yau, P.; Doi, R.H.; Chuang, R.Y. Interactions of opioid and chemokine receptors: oligomerization of mu, kappa, and delta with CCR5 on immune cells. Exp. Cell Res. 2002, 280, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Hogan, C.M.; Hammer, S.M. Host determinants in HIV infection and disease. Part 2: genetic factors and implications for antiretroviral therapeutics. Ann. Intern. Med. 2001, 134, 978–996. [Google Scholar] [PubMed]
- Reynes, J.; Portales, P.; Segondy, M.; Baillat, V.; Andre, P.; Reant, B.; Avinens, O.; Couderc, G.; Benkirane, M.; Clot, J.; Eliaou, J.F.; Corbeau, P. CD4+ T cell surface CCR5 density as a determining factor of virus load in persons infected with human immunodeficiency virus type 1. J. Infect. Dis. 2000, 181, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Butto, S.; Lukwiya, M.; Saresella, M.; Declich, S.; Trabattoni, D.; Pastori, C.; Piconi, S.; Fracasso, C.; Fabiani, M.; Ferrante, P.; Rizzardini, G.; Lopalco, L. Immune activation in Africa is environmentally-driven and is associated with upregulation of CCR5. Italian-Ugandan AIDS Project. Aids 2000, 14, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- de Vries, F.P.; van Der Ende, A.; van Putten, J.P.; Dankert, J. Invasion of primary nasopharyngeal epithelial cells by Neisseria meningitidis is controlled by phase variation of multiple surface antigens. Infect. Immun. 1996, 64, 2998–3006. [Google Scholar] [PubMed]
- Quillent, C.; Oberlin, E.; Braun, J.; Rousset, D.; Gonzalez-Canali, G.; Metais, P.; Montagnier, L.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Beretta, A. HIV-1-resistance phenotype conferred by combination of two separate inherited mutations of CCR5 gene. Lancet 1998, 351, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Lehner, T. The role of CCR5 chemokine ligands and antibodies to CCR5 coreceptors in preventing HIV infection. Trends Immunol. 2002, 23, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Balotta, C.; Bagnarelli, P.; Violin, M.; Ridolfo, A.L.; Zhou, D.; Berlusconi, A.; Corvasce, S.; Corbellino, M.; Clementi, M.; Clerici, M.; Moroni, M.; Galli, M. Homozygous delta 32 deletion of the CCR-5 chemokine receptor gene in an HIV-1-infected patient. Aids 1997, 11, F67–F71. [Google Scholar] [CrossRef] [PubMed]
- Biti, R.; Ffrench, R.; Young, J.; Bennetts, B.; Stewart, G.; Liang, T. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat. Med. 1997, 3, 252–253. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, I.; Meyer, L.; Magierowska, M.; Katlama, C.; Rouzioux, C. HIV-1 infection in an individual homozygous for CCR5 delta 32. Seroco Study Group. Lancet 1997, 349, 1219–1220. [Google Scholar] [CrossRef] [PubMed]
- Gorry, P.R.; Zhang, C.; Wu, S.; Kunstman, K.; Trachtenberg, E.; Phair, J.; Wolinsky, S.; Gabuzda, D. Persistence of dual-tropic HIV-1 in an individual homozygous for the CCR5 Delta 32 allele. Lancet 2002, 359, 1832–1834. [Google Scholar] [CrossRef] [PubMed]
- Naif, H.M.; Cunningham, A.L.; Alali, M.; Li, S.; Nasr, N.; Buhler, M.M.; Schols, D.; de Clercq, E.; Stewart, G. A human immunodeficiency virus type 1 isolate from an infected person homozygous for CCR5Delta32 exhibits dual tropism by infecting macrophages and MT2 cells via CXCR4. J. Virol. 2002, 76, 3114–3124. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, H.W.; Celum, C.; Michael, N.L.; O'Brien, S.; Dean, M.; Carrington, M.; Dondero, D.; Buchbinder, S.P. HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion. J. Acquir. Immune. Defic. Syndr. 2002, 29, 307–313. [Google Scholar] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; Blau, I.W.; Hofmann, W.K.; Thiel, E. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.J.; Ashton, L.J.; Biti, R.A.; Ffrench, R.A.; Bennetts, B.H.; Newcombe, N.R.; Benson, E.M.; Carr, A.; Cooper, D.A.; Kaldor, J.M. Increased frequency of CCR-5 delta 32 heterozygotes among long-term non-progressors with HIV-1 infection. The Australian Long-Term Non-Progressor Study Group. Aids 1997, 11, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Meyer, L.; Magierowska, M.; Hubert, J.B.; Rouzioux, C.; Deveau, C.; Sanson, F.; Debre, P.; Delfraissy, J.F.; Theodorou, I. Early protective effect of CCR-5 delta 32 heterozygosity on HIV-1 disease progression: relationship with viral load. The SEROCO Study Group. Aids 1997, 11, F73–F78. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A. Safety concerns about CCR5 as an antiviral target. Curr. Opin. HIV AIDS 2009, 4, 131–135. [Google Scholar] [CrossRef]
- Moore, J.P.; Klasse, P.J. HIV-1 pathogenesis: the complexities of the CCR5-CCL3L1 complex. Cell Host Microbe 2007, 2, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kurihara, T.; Ryseck, R.P.; Yang, Y.; Ryan, C.; Loy, J.; Warr, G.; Bravo, R. Impaired macrophage function and enhanced T cell-dependent immune response in mice lacking CCR5, the mouse homologue of the major HIV-1 coreceptor. J. Immunol. 1998, 160, 4018–4025. [Google Scholar] [PubMed]
- Glass, W.G.; Lim, J.K.; Cholera, R.; Pletnev, A.G.; Gao, J.L.; Murphy, P.M. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005, 202, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; Petersen, K.; Malmgaard, L.; Mogensen, S.C.; Paludan, S.R. Age-dependent role for CCR5 in antiviral host defense against herpes simplex virus type 2. J. Virol. 2005, 79, 9831–9841. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.; Louie, C.Y.; Glaser, C.; Jean, C.; Johnson, B.; Johnson, H.; McDermott, D.H.; Murphy, P.M. Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J. Infect. Dis. 2008, 197, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.; McHale, M.; Jackson, V.; Penny, M. Assessing theoretical risk and benefit suggested by genetic association studies of CCR5: experience in a drug development programme for maraviroc. Antivir. Ther. 2007, 12, 233–245. [Google Scholar] [PubMed]
- Prahalad, S. Negative association between the chemokine receptor CCR5-Delta32 polymorphism and rheumatoid arthritis: a meta-analysis. Genes Immun. 2006, 7, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Mukai, T.; Gao, P.; Yamaguchi, N.; Ono, S.; Iwaki, H.; Obika, S.; Imanishi, T.; Tsujimura, T.; Hamaoka, T.; Fujiwara, H. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur. J. Immunol. 2002, 32, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Schroder, C.; Pierson 3rd., R.N.; Nguyen, B.N.; Kawka, D.W.; Peterson, L.B.; Wu, G.; Zhang, T.; Springer, M.S.; Siciliano, S.J.; Iliff, S.; Ayala, J.M.; Lu, M.; Mudgett, J.S.; Lyons, K.; Mills, S.G.; Miller, G.G.; Singer, II; Azimzadeh, A.M.; DeMartino, J.A. CCR5 blockade modulates inflammation and alloimmunity in primates. J. Immunol. 2007, 179, 2289–2299. [Google Scholar] [PubMed]
- Berson, J.F.; Doms, R.W. Structure-function studies of the HIV-1 coreceptors. Semin. Immunol. 1998, 10, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Vanderwinden, J.M.; Cihak, J.; Wittamer, V.; Le Poul, E.; Issafras, H.; Stangassinger, M.; Vassart, G.; Marullo, S.; Schlndorff, D.; Parmentier, M.; Mack, M. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol. Biol. Cell 2002, 13, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Locati, M.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. HIV-1 coreceptor activity of CCR5 and its inhibition by chemokines: independence from G protein signaling and importance of coreceptor downmodulation. Virology 1997, 234, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; Davis, C.B.; Peiper, S.C.; Schall, T.J.; Littman, D.R.; Landau, N.R. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; Paxton, W.A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M.; Lalezari, J.; Goodrich, J.; Clumeck, N.; DeJesus, E.; Horban, A.; Nadler, J.; Clotet, B.; Karlsson, A.; Wohlfeiler, M.; Montana, J.B.; McHale, M.; Sullivan, J.; Ridgway, C.; Felstead, S.; Dunne, M.W.; van der Ryst, E.; Mayer, H. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Pope, M. Mucosal dendritic cells and immunodeficiency viruses. J. Infect. Dis. 1999, 179, S427–S430. [Google Scholar] [CrossRef] [PubMed]
- Granelli-Piperno, A.; Delgado, E.; Finkel, V.; Paxton, W.; Steinman, R.M. Immature dendritic cells selectively replicate macrophagetropic (M-tropic) human immunodeficiency virus type 1, while mature cells efficiently transmit both M- and T-tropic virus to T cells. J. Virol. 1998, 72, 2733–2737. [Google Scholar] [PubMed]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; Figdor, C.G.; van Kooyk, Y. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Sodora, D.L.; Koup, R.A.; Paiardini, M.; O'Neil, S.P.; McClure, H.M.; Staprans, S.I.; Feinberg, M.B. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 2003, 18, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Meier-Schellersheim, M.; Sousa, A.E.; Victorino, R.M.; Paul, W.E. CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat. Med. 2002, 8, 319–323. [Google Scholar] [CrossRef]
- Berkowitz, R.D.; Alexander, S.; McCune, J.M. Causal relationships between HIV-1 coreceptor utilization, tropism, and pathogenesis in human thymus. AIDS Res. Hum. Retroviruses 2000, 16, 1039–1045. [Google Scholar] [PubMed]
- Correa, R.; Munoz-Fernandez, M.A. Viral phenotype affects the thymic production of new T cells in HIV-1-infected children. Aids 2001, 15, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Stuart, J.W.; Otto, S.A.; Borleffs, J.C.; Boucher, C.A.; de Boer, R.J.; Miedema, F.; Hamann, D. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 2000, 95, 249–255. [Google Scholar] [PubMed]
- van Rij, R.P.; Hazenberg, M.D.; van Benthem, B.H.; Otto, S.A.; Prins, M.; Miedema, F.; Schuitemaker, H. Early viral load and CD4+ T cell count, but not percentage of CCR5+ or CXCR4+ CD4+ T cells, are associated with R5-to-X4 HIV type 1 virus evolution. AIDS Res. Hum. Retroviruses 2003, 19, 389–398. [Google Scholar] [PubMed]
- Moore, J.P.; Kitchen, S.G.; Pugach, P.; Zack, J.A. The CCR5 and CXCR4 coreceptors--central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retroviruses 2004, 20, 111–126. [Google Scholar] [PubMed]
- Margolis, L.; Shattock, R. Selective transmission of CCR5-utilizing HIV-1: the 'gatekeeper' problem resolved? Nat. Rev. Microbiol. 2006, 4, 312–317. [Google Scholar] [CrossRef]
- Bomsel, M.; Alfsen, A. Entry of viruses through the epithelial barrier: pathogenic trickery. Nat. Rev. Mol. Cell Biol. 2003, 4, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J.; Shattock, R.; Moore, J.P. Antiretroviral drug-based microbicides to prevent HIV-1 sexual transmission. Annu. Rev. Med. 2008, 59, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Bourinbaiar, A.S.; Metadilogkul, O.; Jirathitikal, V. Mucosal AIDS vaccines. Viral Immunol. 2003, 16, 427–445. [Google Scholar] [PubMed]
- Lehner, T. Innate and adaptive mucosal immunity in protection against HIV infection. Vaccine 2003, 21, S68–S76. [Google Scholar] [CrossRef] [PubMed]
- Verani, A.; Lusso, P. Chemokines as natural HIV antagonists. Curr. Mol. Med. 2002, 2, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Neutra, M.R.; Kozlowski, P.A. Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; Littman, D.R.; Reinecker, H.C. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; de Jong, E.C.; Baldwin, C.E.; Schuitemaker, J.H.; Kapsenberg, M.L.; Berkhout, B. Differential transmission of human immunodeficiency virus type 1 by distinct subsets of effector dendritic cells. J. Virol. 2002, 76, 7812–7821. [Google Scholar] [CrossRef] [PubMed]
- Hladik, F.; McElrath, M.J. Setting the stage: host invasion by HIV. Nat. Rev. Immunol. 2008, 8, 447–457. [Google Scholar] [CrossRef]
- Mascola, J.R.; Frankel, S.S.; Broliden, K. HIV-1 entry at the mucosal surface: role of antibodies in protection. Aids 2000, 14, S167–S174. [Google Scholar] [PubMed]
- Bouhlal, H.; Latry, V.; Requena, M.; Aubry, S.; Kaveri, S.V.; Kazatchkine, M.D.; Belec, L.; Hocini, H. Natural antibodies to CCR5 from breast milk block infection of macrophages and dendritic cells with primary R5-tropic HIV-1. J. Immunol. 2005, 174, 7202–7209. [Google Scholar] [PubMed]
- Clerici, M. Immune activation in Africa is environmentally-driven and is associated with upregulation of CCR5. Italian-Ugandan AIDS Project . AIDS 2000, 14, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Barassi, C.; Soprana, E.; Pastori, C.; Longhi, R.; Buratti, E.; Lillo, F.; Marenzi, C.; Lazzarin, A.; Siccardi, A.G.; Lopalco, L. Induction of murine mucosal CCR5-reactive antibodies as an anti-human immunodeficiency virus strategy. J. Virol. 2005, 79, 6848–6858. [Google Scholar] [CrossRef] [PubMed]
- Barassi, C.; Lazzarin, A.; Lopalco, L. CCR5-specific mucosal IgA in saliva and genital fluids of HIV-exposed seronegative subjects. Blood 2004, 104, 2205–2206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Underwood, J.; Vaughan, R.; Harmer, A.; Doyle, C.; Lehner, T. Allo-immunization elicits CCR5 antibodies, SDF-1 chemokines, and CD8-suppressor factors that inhibit transmission of R5 and X4 HIV-1 in women. Clin. Exp. Immunol. 2002, 129, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Stott, E.J. Anti-cell antibody in macaques. Nature 1991, 353, 393. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Lopalco, L. Is autoimmunity a component of natural immunity to HIV? Curr. HIV Res. 2006, 4, 177–190. [Google Scholar] [CrossRef]
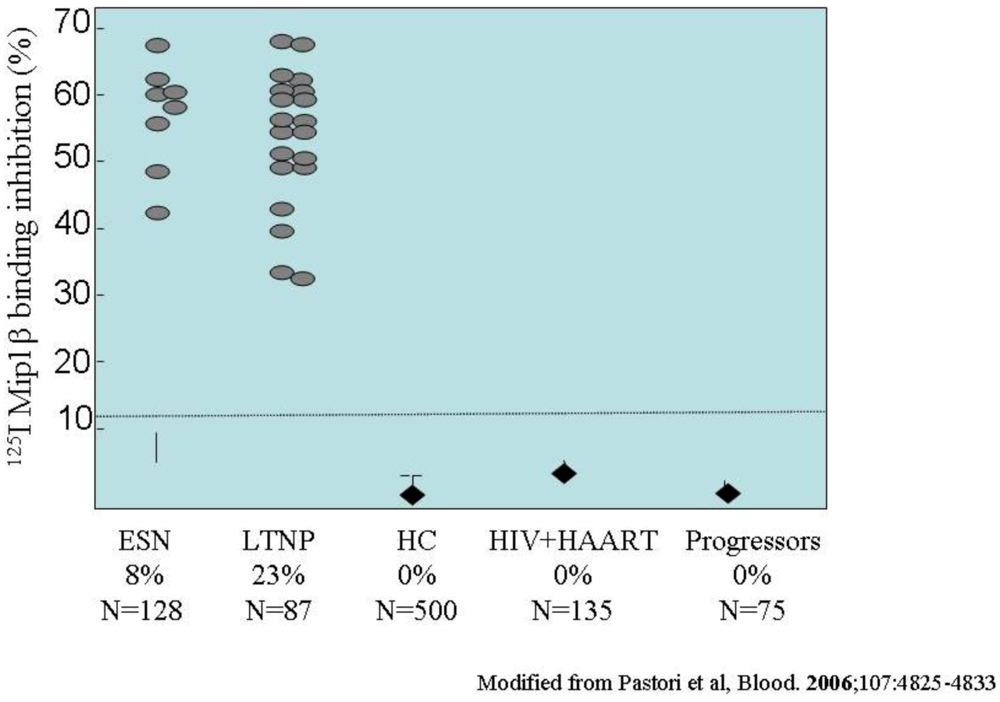
- Pastori, C.; Weiser, B.; Barassi, C.; Uberti-Foppa, C.; Ghezzi, S.; Longhi, R.; Calori, G.; Burger, H.; Kemal, K.; Poli, G.; Lazzarin, A.; Lopalco, L. Long-lasting CCR5 internalization by antibodies in a subset of long-term nonprogressors: a possible protective effect against disease progression. Blood 2006, 107, 4825–4833. [Google Scholar] [CrossRef] [PubMed]
- Oxman, M.N. Herpes zoster pathogenesis and cell-mediated immunity and immunosenescence. J. Am. Osteopath. Assoc. 2009, 109, S13–S17. [Google Scholar] [PubMed]
- Wolinsky, S.M.; Veazey, R.S.; Kunstman, K.J.; Klasse, P.J.; Dufour, J.; Marozsan, A.J.; Springer, M.S.; Moore, J.P. Effect of a CCR5 inhibitor on viral loads in macaques dual-infected with R5 and X4 primate immunodeficiency viruses. Virology 2004, 328, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Kuritzkes, D.R. A piece de resistance: how HIV-1 escapes small molecule CCR5 inhibitors. Curr. Opin. HIV AIDS 2009, 4, 118–124. [Google Scholar] [CrossRef]
- Olson, W.C.; Jacobson, J.M. CCR5 monoclonal antibodies for HIV-1 therapy. Curr. Opin. HIV AIDS 2009, 4, 104–111. [Google Scholar] [CrossRef]
- Heredia, A.; Gilliam, B.; Latinovic, O.; Le, N.; Bamba, D.; Devico, A.; Melikyan, G.B.; Gallo, R.C.; Redfield, R.R. Rapamycin reduces CCR5 density levels on CD4 T cells, and this effect results in potentiation of enfuvirtide (T-20) against R5 strains of human immunodeficiency virus type 1 in vitro. Antimicrob. Agents Chemother. 2007, 51, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Nabatov, A.A.; Pollakis, G.; Linnemann, T.; Paxton, W.A.; de Baar, M.P. Statins disrupt CCR5 and RANTES expression levels in CD4+ T lymphocytes in vitro and preferentially decrease infection of R5 versus X4 HIV-1. PLoS One 2007, 2, e470. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.F.; An, D.S.; Chen, I.S.; Baltimore, D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. USA 2003, 100, 183–188. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Serrano, F.; Llorente, M.; Abad, J.L.; Garcia-Ortiz, M.J.; Bernad, A. A hammerhead ribozyme targeted to the human chemokine receptor CCR5. Biochem. Biophys. Res. Commun. 1998, 251, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Steinberger, P.; Andris-Widhopf, J.; Buhler, B.; Torbett, B.E.; Barbas 3rd., C.F. 3rd Functional deletion of the CCR5 receptor by intracellular immunization produces cells that are refractory to CCR5-dependent HIV-1 infection and cell fusion. Proc. Natl. Acad. Sci. USA 2000, 97, 805–810. [Google Scholar] [CrossRef]
- Yang, A.G.; Bai, X.; Huang, X.F.; Yao, C.; Chen, S. Phenotypic knockout of HIV type 1 chemokine coreceptor CCR-5 by intrakines as potential therapeutic approach for HIV-1 infection. Proc. Natl. Acad. Sci. USA 1997, 94, 11567–11572. [Google Scholar] [CrossRef]
- Tsibris, A.M.; Sagar, M.; Gulick, R.M.; Su, Z.; Hughes, M.; Greaves, W.; Subramanian, M.; Flexner, C.; Giguel, F.; Leopold, K.E.; Coakley, E.; Kuritzkes, D.R. In vivo emergence of vicriviroc resistance in a human immunodeficiency virus type 1 subtype C-infected subject. J. Virol. 2008, 82, 8210–8214. [Google Scholar] [CrossRef] [PubMed]
- Ogert, R.A.; Wojcik, L.; Buontempo, C.; Ba, L.; Buontempo, P.; Ralston, R.; Strizki, J.; Howe, J.A. Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology 2008, 373, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E. How HIV changes its tropism: evolution and adaptation? Curr. Opin. HIV AIDS 2009, 4, 125–130. [Google Scholar] [CrossRef]
- Borggren, M.; Repits, J.; Kuylenstierna, C.; Sterjovski, J.; Churchill, M.J.; Purcell, D.F.; Karlsson, A.; Albert, J.; Gorry, P.R.; Jansson, M. Evolution of DC-SIGN use revealed by fitness studies of R5 HIV-1 variants emerging during AIDS progression. Retrovirology 2008, 5, 28. [Google Scholar] [CrossRef]
- Repits, J.; Sterjovski, J.; Badia-Martinez, D.; Mild, M.; Gray, L.; Churchill, M.J.; Purcell, D.F.; Karlsson, A.; Albert, J.; Fenyo, E.M.; Achour, A.; Gorry, P.R.; Jansson, M. Primary HIV-1 R5 isolates from end-stage disease display enhanced viral fitness in parallel with increased gp120 net charge. Virology 2008, 379, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Low, A.J.; Marchant, D.; Brumme, C.J.; Brumme, Z.L.; Dong, W.; Sing, T.; Hogg, R.S.; Montaner, J.S.; Gill, V.; Cheung, P.K.; Harrigan, P.R. CD4-dependent characteristics of coreceptor use and HIV type 1 V3 sequence in a large population of therapy-naive individuals. AIDS Res. Hum. Retroviruses 2008, 24, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Eshleman, S.H.; Toma, J.; Fransen, S.; Stawiski, E.; Paxinos, E.E.; Whitcomb, J.M.; Young, A.M.; Donnell, D.; Mmiro, F.; Musoke, P.; Guay, L.A.; Jackson, J.B.; Parkin, N.T.; Petropoulos, C.J. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J. Virol. 2007, 81, 7885–7893. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Wrin, T.; Little, S.J.; Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 2003, 100, 4144–4149. [Google Scholar] [CrossRef]
- Pugach, P.; Ketas, T.J.; Michael, E.; Moore, J.P. Neutralizing antibody and anti-retroviral drug sensitivities of HIV-1 isolates resistant to small molecule CCR5 inhibitors. Virology 2008, 377, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Trkola, A.; Ketas, T.J.; Nagashima, K.A.; Zhao, L.; Cilliers, T.; Morris, L.; Moore, J.P.; Maddon, P.J.; Olson, W.C. Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J. Virol. 2001, 75, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.M.; Saag, M.S.; Thompson, M.A.; Fischl, M.A.; Liporace, R.; Reichman, R.C.; Redfield, R.R.; Fichtenbaum, C.J.; Zingman, B.S.; Patel, M.C.; Murga, J.D.; Pemrick, S.M.; D'Ambrosio, P.; Michael, M.; Kroger, H.; Ly, H.; Rotshteyn, Y.; Buice, R.; Morris, S.A.; Stavola, J.J.; Maddon, P.J.; Kremer, A.B.; Olson, W.C. Antiviral activity of single-dose PRO 140, a CCR5 monoclonal antibody, in HIV-infected adults. J. Infect. Dis. 2008, 198, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.; Yadavalli, G.K.; Para, M.; Richmond, G.; Dejesus, E.; Brown, S.J.; Cai, W.; Chen, C.; Zhong, J.; Novello, L.A.; Lederman, M.M.; Subramanian, G.M. Safety, pharmacokinetics, and antiviral activity of HGS004, a novel fully human IgG4 monoclonal antibody against CCR5, in HIV-1-infected patients. J. Infect. Dis. 2008, 197, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, S.; La Seta-Catamancio, S.; Kurtagic, S.; Galazzi, M.; Arienti, D.; Trabattoni, D.; Wilken, J.; Thompson, D.A.; Offord, R.E.; Galli, M.; Clerici, M. Aminooxypentane-RANTES, an inhibitor of R5 human immunodeficiency virus type 1, increases the interferon gamma to interleukin 10 ratio without impairing cellular proliferation. AIDS Res. Hum. Retroviruses 1999, 15, 861–867. [Google Scholar] [PubMed]
- Mack, M.; Luckow, B.; Nelson, P.J.; Cihak, J.; Simmons, G.; Clapham, P.R.; Signoret, N.; Marsh, M.; Stangassinger, M.; Borlat, F.; Wells, T.N.; Schlondorff, D.; Proudfoot, A.E. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J. Exp. Med. 1998, 187, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, H.; Cerini, F.; Escola, J.M.; Kuenzi, G.; Melotti, A.; Offord, R.; Rossitto-Borlat, I.; Nedellec, R.; Salkowitz, J.; Gorochov, G.; Mosier, D.; Hartley, O. Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. USA 2008, 105, 17706–17711. [Google Scholar] [CrossRef]
- Veazey, R.S.; Ling, B.; Green, L.C.; Ribka, E.P.; Lifson, J.D.; Piatak Jr., M.; Lederman, M.M.; Mosier, D.; Offord, R.; Hartley, O. Topically applied recombinant chemokine analogues fully protect macaques from vaginal simian-human immunodeficiency virus challenge. J. Infect. Dis. 2009, 199, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E.; Picchio, G.R.; Gulizia, R.J.; Sabbe, R.; Poignard, P.; Picard, L.; Offord, R.E.; Thompson, D.A.; Wilken, J. Highly potent RANTES analogues either prevent CCR5-using human immunodeficiency virus type 1 infection in vivo or rapidly select for CXCR4-using variants. J. Virol. 1999, 73, 3544–3550. [Google Scholar] [PubMed]
- Lopalco, L.; Pastori, C.; Cosma, A.; Burastero, S.E.; Capiluppi, B.; Boeri, E.; Beretta, A.; Lazzarin, A.; Siccardi, A.G. Anti-cell antibodies in exposed seronegative individuals with HIV type 1-neutralizing activity. AIDS Res. Hum. Retroviruses 2000, 16, 109–115. [Google Scholar] [PubMed]
- Chain, B.M.; Noursadeghi, M.; Gardener, M.; Tsang, J.; Wright, E. HIV blocking antibodies following immunisation with chimaeric peptides coding a short N-terminal sequence of the CCR5 receptor. Vaccine 2008, 26, 5752–5759. [Google Scholar] [CrossRef] [PubMed]
- Devito, C.; Zuber, B.; Schroder, U.; Benthin, R.; Okuda, K.; Broliden, K.; Wahren, B.; Hinkula, J. Intranasal HIV-1-gp160-DNA/gp41 peptide prime-boost immunization regimen in mice results in long-term HIV-1 neutralizing humoral mucosal and systemic immunity. J. Immunol. 2004, 173, 7078–7089. [Google Scholar] [PubMed]
- Buratti, E.; McLain, L.; Tisminetzky, S.; Cleveland, S.M.; Dimmock, N.J.; Baralle, F.E. The neutralizing antibody response against a conserved region of human immunodeficiency virus type 1 gp41 (amino acid residues 731-752) is uniquely directed against a conformational epitope. J. Gen Virol. 1998, 79, 2709–2716. [Google Scholar] [PubMed]
- Pastori, C.; Clivio, A.; Diomede, L.; Consonni, R.; De Mori, G.M.; Longhi, R.; Colombo, G.; Lopalco, L. Two amino acid substitutions within the first external loop of CCR5 induce human immunodeficiency virus-blocking antibodies in mice and chickens. J. Virol. 2008, 82, 4125–4134. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, B.; Lowy, D.R.; Schiller, J.T. Induction of autoantibodies to mouse CCR5 with recombinant papillomavirus particles. Proc. Natl. Acad. Sci. USA 1999, 96, 2373–2378. [Google Scholar] [CrossRef]
- Bogers, W.M.; Bergmeier, L.A.; Ma, J.; Oostermeijer, H.; Wang, Y.; Kelly, C.G.; Ten Haaft, P.; Singh, M.; Heeney, J.L.; Lehner, T. A novel HIV-CCR5 receptor vaccine strategy in the control of mucosal SIV/HIV infection. Aids 2004, 18, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Misumi, S.; Nakayama, D.; Kusaba, M.; Iiboshi, T.; Mukai, R.; Tachibana, K.; Nakasone, T.; Umeda, M.; Shibata, H.; Endo, M.; Takamune, N.; Shoji, S. Effects of immunization with CCR5-based cycloimmunogen on simian/HIVSF162P3 challenge. J. Immunol. 2006, 176, 463–471. [Google Scholar] [PubMed]
- Fauci, A. Pathogenesis of HIV disease: opportunities for new prevention interventions. Clinical Infectious Diseases 2007, 45, S206–S212. [Google Scholar] [CrossRef] [PubMed]
- Paxton, W.A.; Kang, S. Chemokine receptor allelic polymorphisms: relationships to HIV resistance and disease progression. Semin. Immunol. 1998, 10, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Paxton, W.A.; Kassam, N.; Ruffing, N.; Rottman, J.B.; Sullivan, N.; Choe, H.; Sodroski, J.; Newman, W.; Koup, R.A.; Mackay, C.R. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 1997, 185, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Fatkenheuer, G.; Nelson, M.; Lazzarin, A.; Konourina, I.; Hoepelman, A.I.; Lampiris, H.; Hirschel, B.; Tebas, P.; Raffi, F.; Trottier, B.; Bellos, N.; Saag, M.; Cooper, D.A.; Westby, M.; Tawadrous, M.; Sullivan, J.F.; Ridgway, C.; Dunne, M.W.; Felstead, S.; Mayer, H.; van der Ryst, E. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 2008, 359, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Lopalco, L. CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses 2010, 2, 574-600. https://doi.org/10.3390/v2020574
Lopalco L. CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses. 2010; 2(2):574-600. https://doi.org/10.3390/v2020574
Chicago/Turabian StyleLopalco, Lucia. 2010. "CCR5: From Natural Resistance to a New Anti-HIV Strategy" Viruses 2, no. 2: 574-600. https://doi.org/10.3390/v2020574
APA StyleLopalco, L. (2010). CCR5: From Natural Resistance to a New Anti-HIV Strategy. Viruses, 2(2), 574-600. https://doi.org/10.3390/v2020574