Role of Lymphocytic Choriomeningitis Virus (LCMV) in Understanding Viral Immunology: Past, Present and Future

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
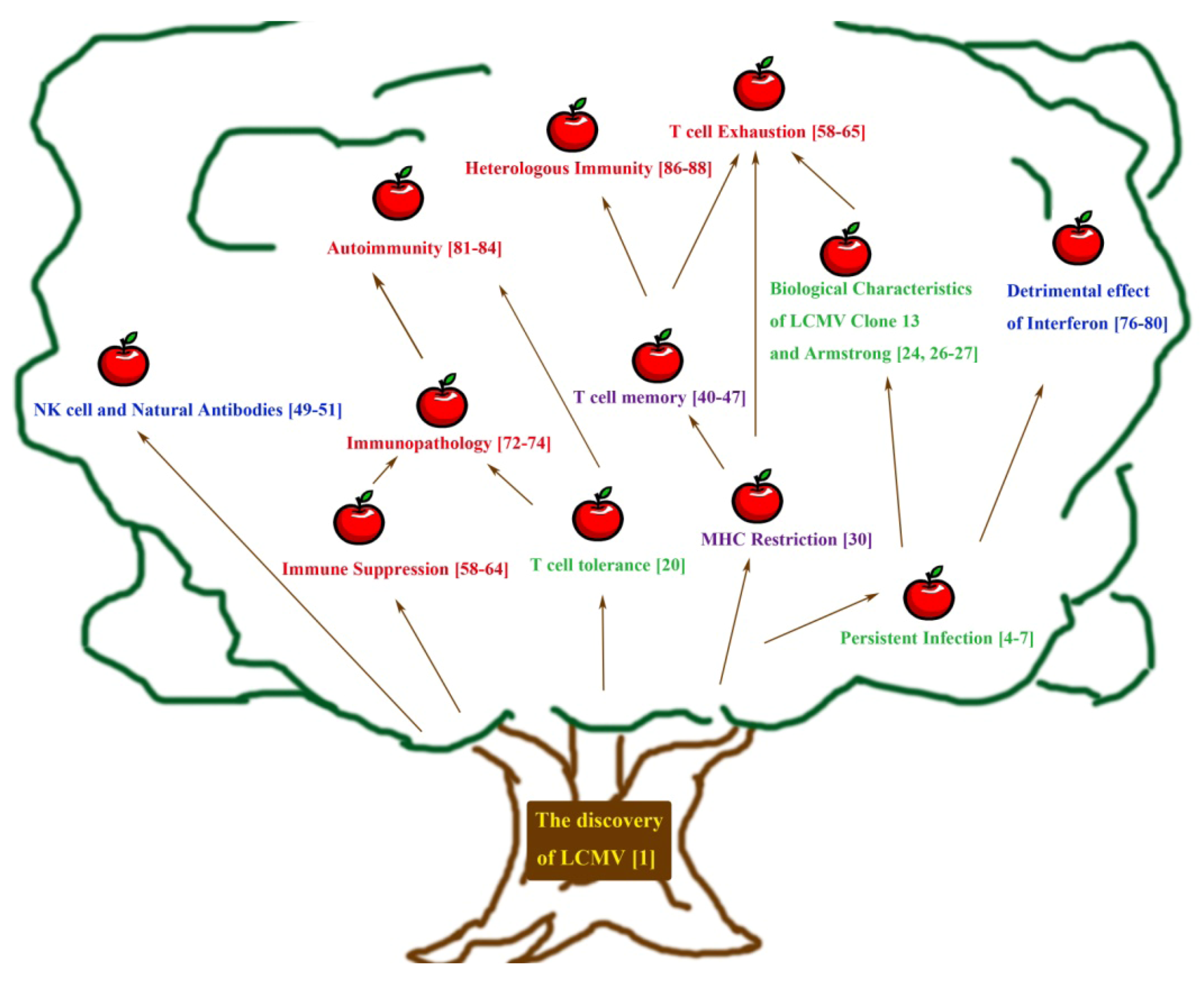
:1. Introduction
2. Early Approaches towards Understanding Persistent Infection and Its Immune Response
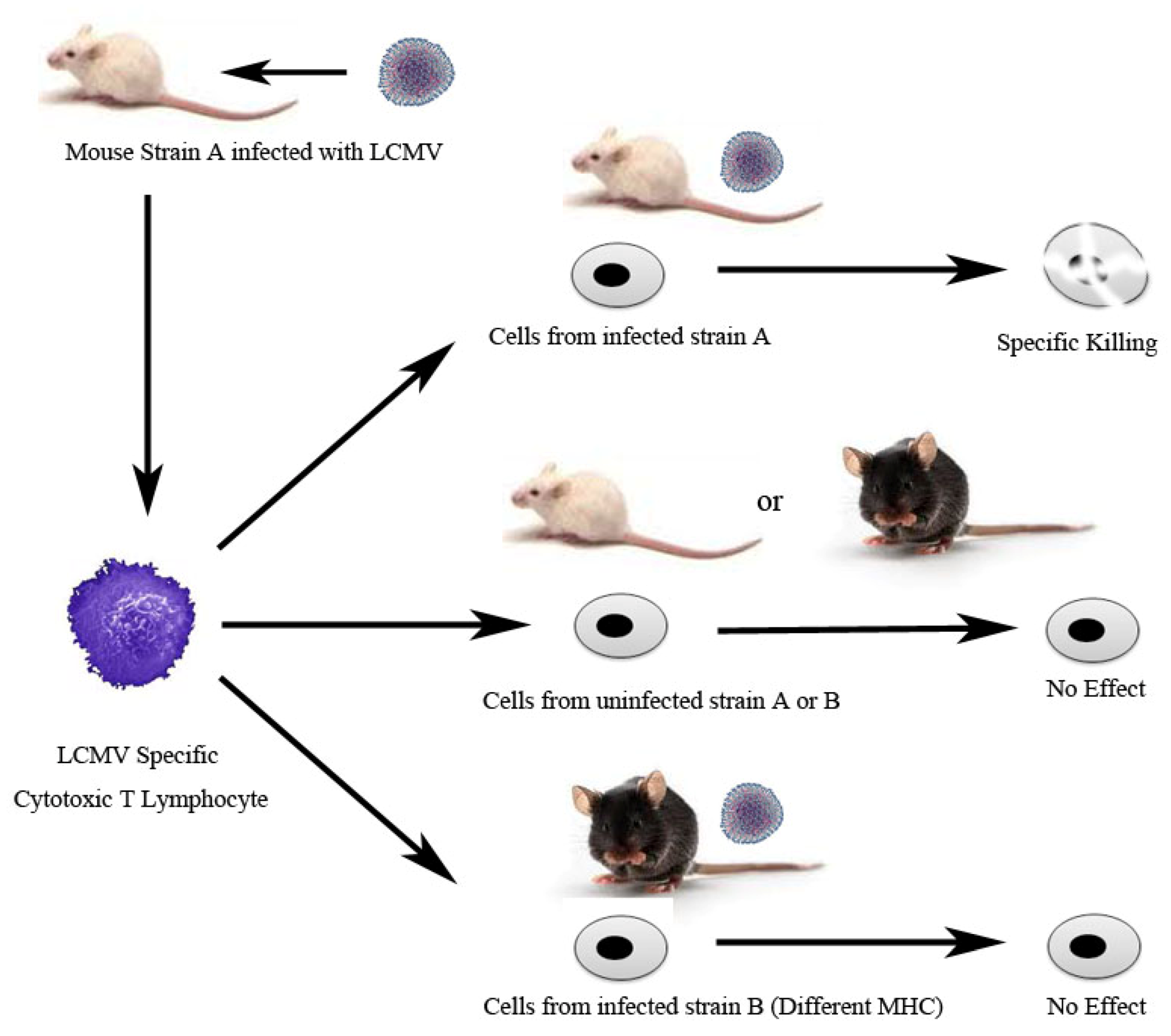
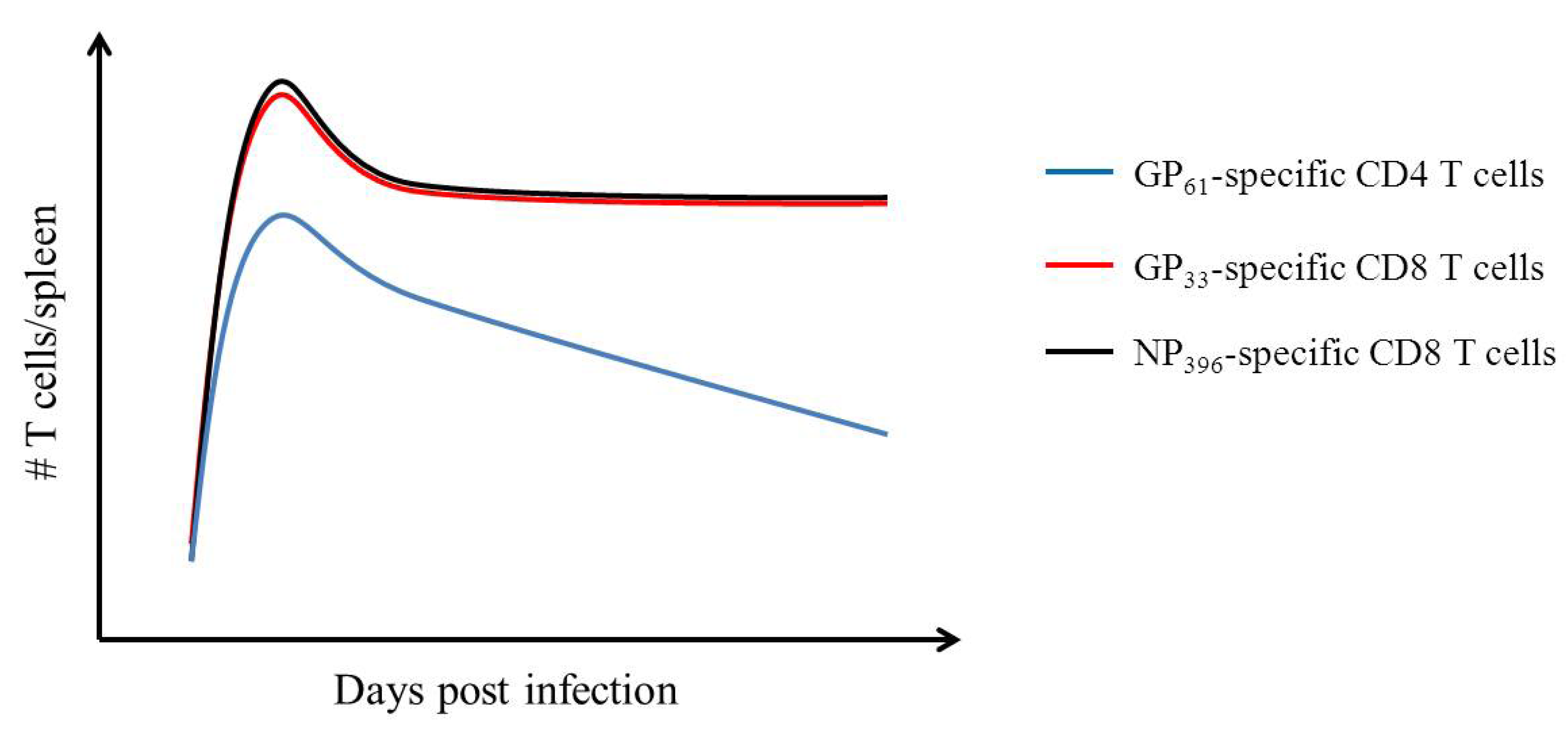
3. Adaptive Immunity: Understanding the T Cell Mediated Immune Response Using LCMV
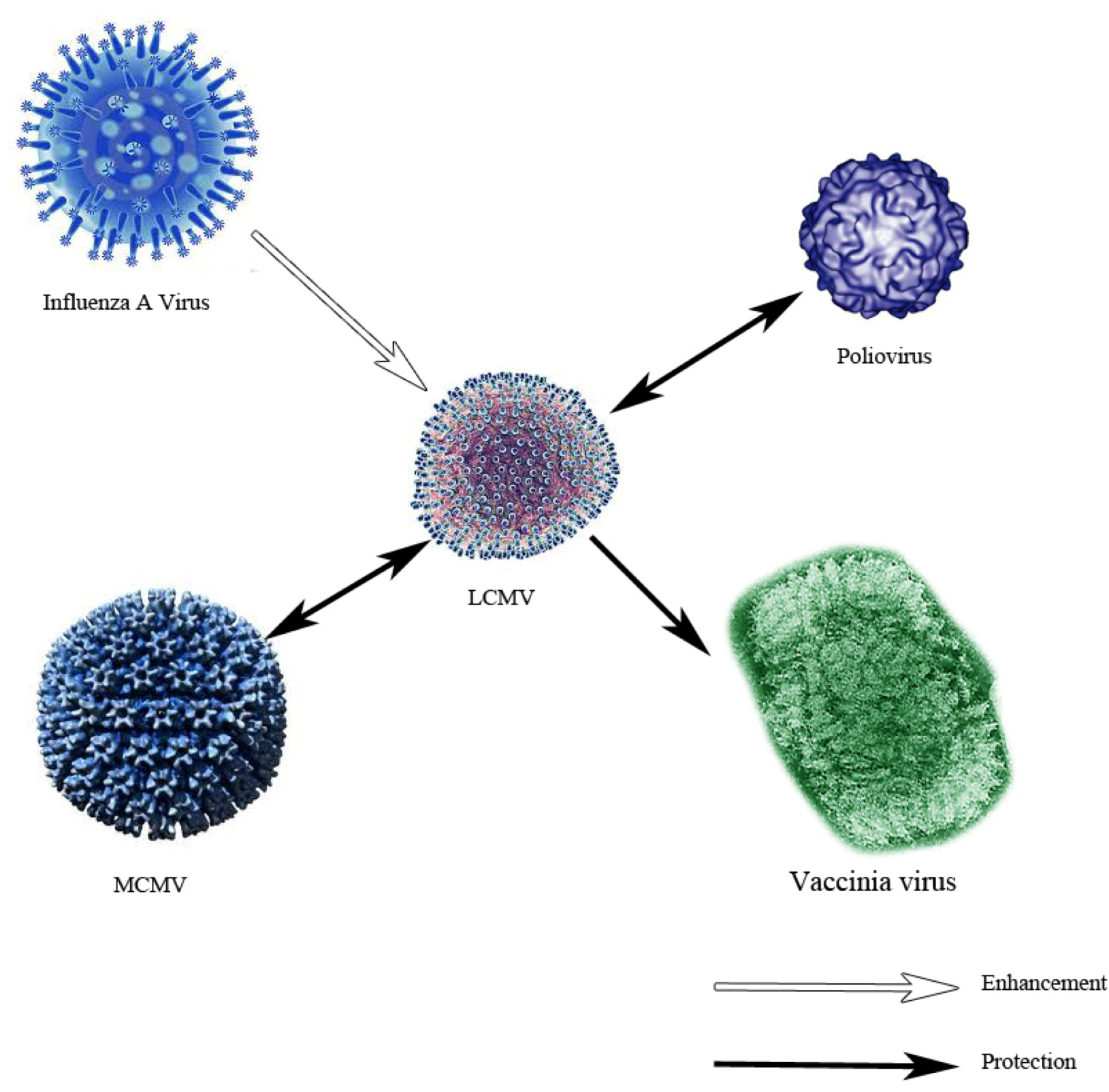
4. Understanding Innate Immunity: Contributions from the LCMV System
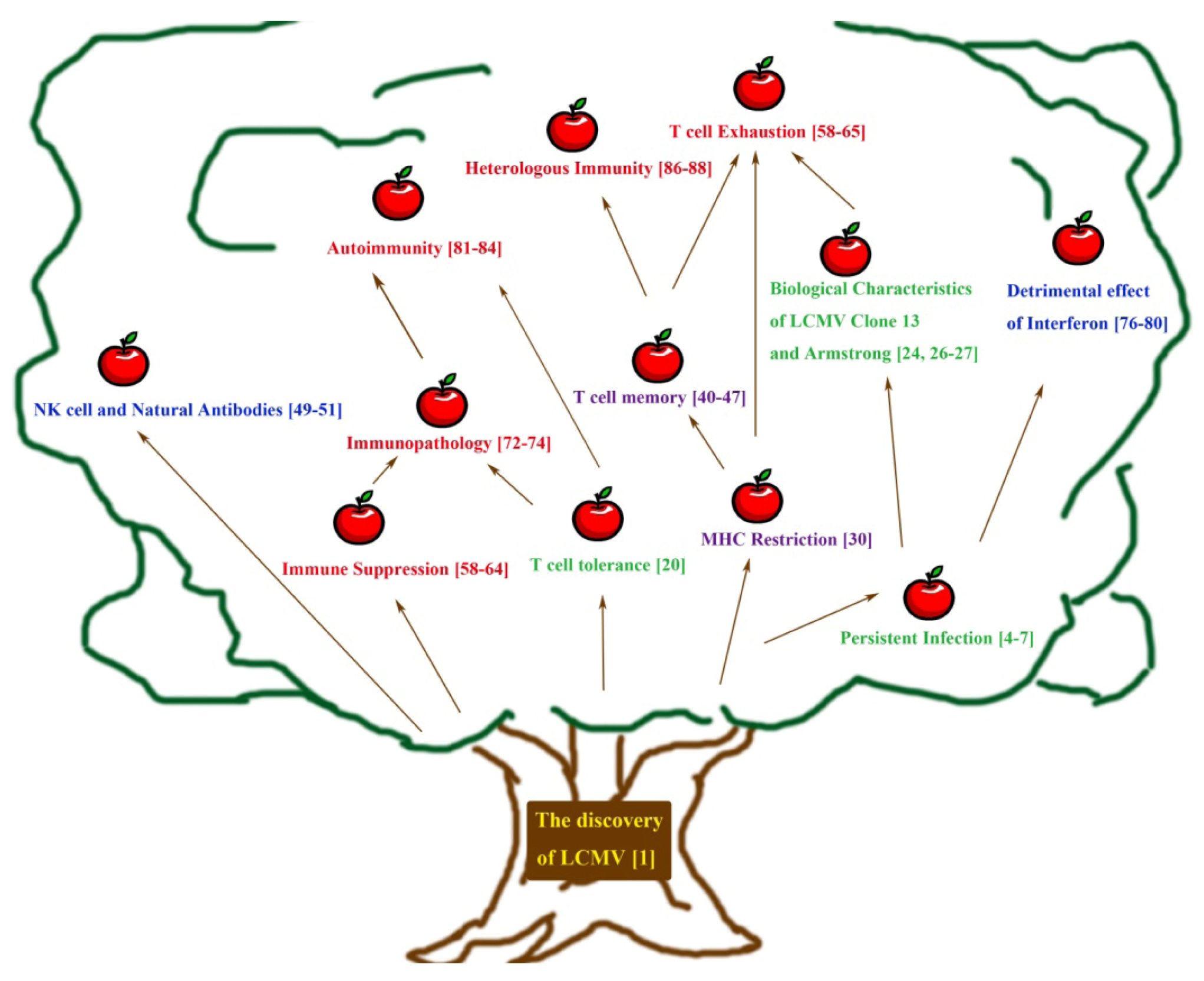
5. Immune Dysfunction as Observed Using the LCMV Model — Immunosuppression, Immune Exhaustion, Immunopathology, Autoimmunity and Heterologous Immunity
6. From the Present and Onwards
Conflict of Interest
Acknowledgements
References and Notes
- Muckenfuss, R.S.; Armstrong, C.; Webster, L. Etiology of the 1933 epidemic of encephalitis. J. Am. Med. Assoc. 1934, 103, 731. [Google Scholar] [CrossRef]
- Beeman, E.A. Charles Armstrong MD: A Biography; National Institutes of Health: Bethesda, MD, USA, 2007. [Google Scholar]
- Luby, J.P. St. Louis encephalitis. Epidemiol. Rev. 1979, 1, 55–73. [Google Scholar]
- Traub, E. An epidemic in a mouse colony due to the virus of acute lymphocytic choriomeningitis. J. Exp. Med. 1936, 63, 533–546. [Google Scholar] [CrossRef]
- Traub, E. Persistence of lymphocytic choriomeningitis virus in immune animals and its relation to immunity. J. Exp. Med. 1936, 63, 847–861. [Google Scholar] [CrossRef]
- Rivers, T.M.; Scott, T.F. Meningitis in man caused by a filterable virus: II. Identification of the etiological agent. J. Exp. Med. 1936, 63, 415–432. [Google Scholar] [CrossRef]
- Traub, E. The epidemiology of lymphocytic choriomeningitis in white mice. J. Exp. Med. 1936, 64, 183–200. [Google Scholar] [CrossRef]
- Burnet, F.M.; Fenner, F. The Production of Antibodies, 2nd ed; Macmillan: Melbourne, Australia, 1949; pp. viii, 142. [Google Scholar]
- Weigand, H.; Hotchin, J. Studies of lymphocytic choriomeningitis in mice. J. Immunol. 1961, 86, 401. [Google Scholar]
- Oldstone, M.B.; Dixon, F.J. Lymphocytic choriomeningitis: Production of antibody by "tolerant" infected mice. Science 1967, 158, 1193–1195. [Google Scholar]
- Hotchin, J. The contamination of laboratory animals with lymphocytic choriomeningitis virus. Am. J. Pathol. 1971, 64, 747–769. [Google Scholar]
- Benson, L.; Hotchin, J. Antibody formation in persistent tolerant infection with lymphocytic choriomeningitis virus. Nature 1969, 222, 1045–1047. [Google Scholar] [CrossRef]
- Rowe, W. Studies on pathogenesis and immunity in lymphocytic choriomeningitis infection of the mouse. Rev. Rep. Naval Med. Res. Inst. 1954, 12, 167–220. [Google Scholar]
- Hotchin, J.; Benson, L.M.; Seamer, J. Factors affecting the induction of persistent tolerant infection of newborn mice with lymphocytic choriomeningitis. Virology 1962, 18, 71–78. [Google Scholar] [CrossRef]
- Hotchin, J. The biology of lymphocytic choriomeningitis infection: Virus-induced immune disease. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 479–499. [Google Scholar] [CrossRef]
- Hotchin, J.; Benson, L. The pathogenesis of lymphocytic choriomeningitis in mice: The effects of different inoculation routes and the footpad response. J. Immunol. 1963, 91, 460–468. [Google Scholar]
- Oldstone, M.B.; Dixon, F.J. Susceptibility of different mouse strains to lymphocytic choriomeningitis virus. J. Immunol. 1968, 100, 355–357. [Google Scholar]
- Lehmann-Grube, F.; Slenczka, W.; Tees, R. A persistent and inapparent infection of L cells with the virus of lymphocytic choriomeningitis. J. Gen. Virol. 1969, 5, 63–81. [Google Scholar] [CrossRef]
- Lehmann-Grube, F. Lymphocytic choriomeningitis in the mouse. I. Growth in the brain. Arch Gesamte Virusforsch 1964, 14, 344–350. [Google Scholar] [CrossRef]
- Volkert, M.; Bro-Jorgensen, K.; Marker, O. Persistent LCM virus infection in the mouse. Immunity and tolerance. Bull. World Health Organ. 1975, 52, 471–478. [Google Scholar]
- Oldstone, M.B.; Buchmeier, M.J.; Doyle, M.V.; Tishon, A. Virus-induced immune complex disease: Specific anti-viral antibody and C1q binding material in the circulation during persistent lymphocytic choriomeningitis virus infection. J. Immunol. 1980, 124, 831–838. [Google Scholar]
- Oldstone, M.B.; Tishon, A.; Buchmeier, M.J. Virus-induced immune complex disease: Genetic control of C1q binding complexes in the circulation of mice persistently infected with lymphocytic choriomeningitis virus. J. Immunol. 1983, 130, 912–918. [Google Scholar]
- Tishon, A.; Salmi, A.; Ahmed, R.; Oldstone, M.B. Role of viral strains and host genes in determining levels of immune complexes in a model system: Implications for HIV infection. AIDS Res. Hum. Retrovir. 1991, 7, 963–969. [Google Scholar] [CrossRef]
- Ahmed, R.; Salmi, A.; Butler, L.D.; Chiller, J.M.; Oldstone, M.B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 1984, 160, 521–540. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar]
- Salvato, M.; Borrow, P.; Shimomaye, E.; Oldstone, M.B. Molecular basis of viral persistence: A single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J. Virol. 1991, 65, 1863–1869. [Google Scholar]
- Matloubian, M.; Kolhekar, S.R.; Somasundaram, T.; Ahmed, R. Molecular determinants of macrophage tropism and viral persistence: Importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J. Virol. 1993, 67, 7340–7349. [Google Scholar]
- Balint, A.; Farsang, A.; Zadori, Z.; Hornyak, A.; Dencso, L.; Almazan, F.; Enjuanes, L.; Belak, S. Molecular characterization of feline infectious peritonitis virus strain DF-2 and studies on the role of ORF3abc in viral cell tropism. J. Virol. 2012, 86, 6258–6267. [Google Scholar] [CrossRef]
- Tishon, A.; Oldstone, M.B. Persistent virus infection associated with chemical manifestations of diabetes. II. Role of viral strain, environmental insult, and host genetics. Am. J. Pathol. 1987, 126, 61–72. [Google Scholar]
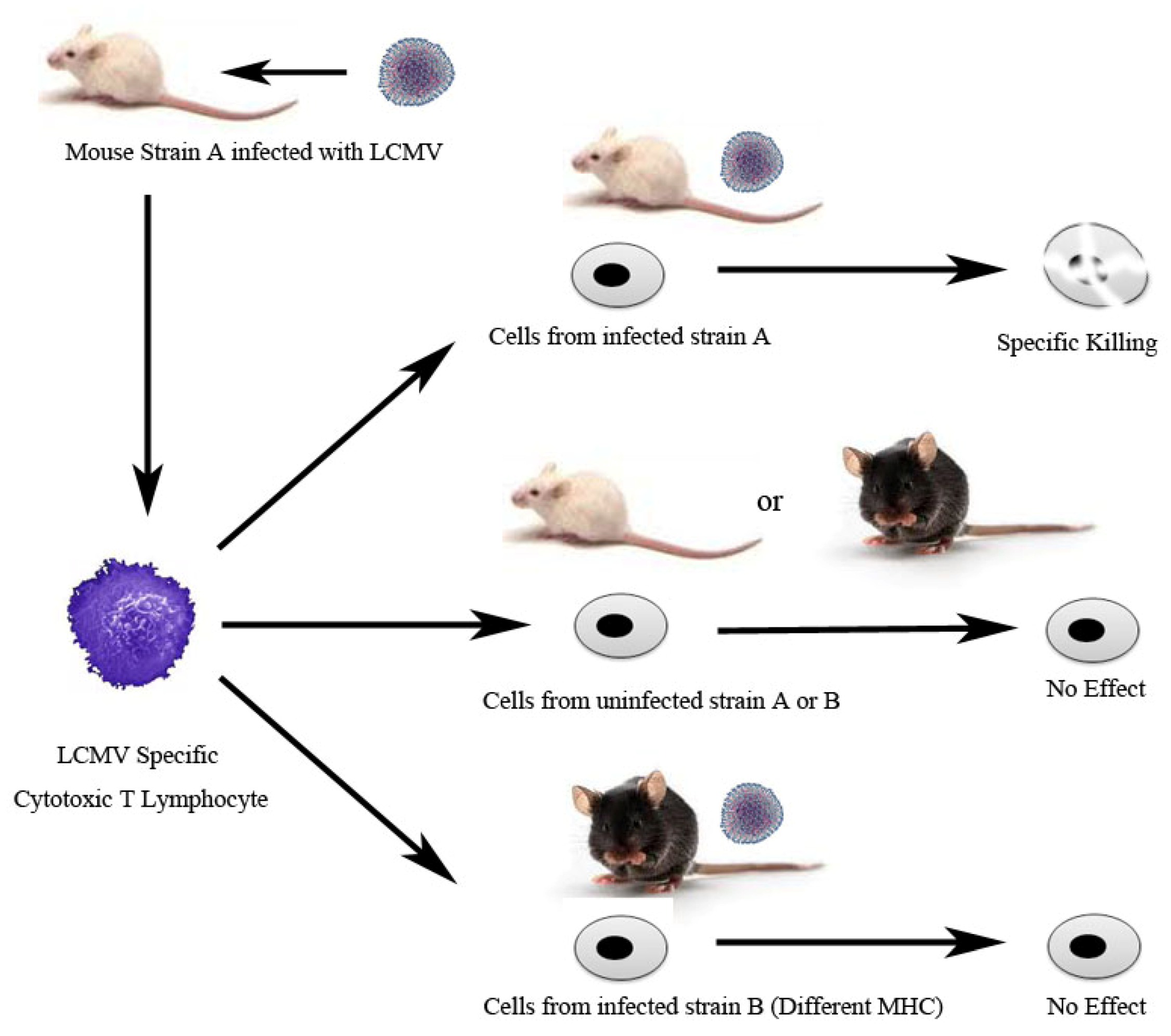
- Zinkernagel, R.M.; Doherty, P.C. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 1974, 248, 701–702. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Doherty, P.C. Immunological surveillance against altered self components by sensitised T lymphocytes in lymphocytic choriomeningitis. Nature 1974, 251, 547–548. [Google Scholar] [CrossRef]
- Doherty, P.C.; Zinkernagel, R.M. H-2 compatibility is required for T-cell-mediated lysis of target cells infected with lymphocytic choriomeningitis virus. J. Exp. Med. 1975, 141, 502–507. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Doherty, P.C. H-2 compatability requirement for T-cell-mediated lysis of target cells infected with lymphocytic choriomeningitis virus. Different cytotoxic T-cell specificities are associated with structures coded for in H-2K or H-2D. J. Exp. Med. 1975, 141, 1427–1436. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; Welsh, R.M.; Dutko, F.J.; Oldstone, M.B. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv. Immunol. 1980, 30, 275–331. [Google Scholar] [CrossRef]
- Byrne, J.A.; Ahmed, R.; Oldstone, M.B. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus. I. Generation and recognition of virus strains and H-2b mutants. J. Immunol. 1984, 133, 433–439. [Google Scholar]
- Masson, D.; Tschopp, J. Isolation of a lytic, pore-forming protein (perforin) from cytolytic T-lymphocytes. J. Biol. Chem. 1985, 260, 9069–9072. [Google Scholar]
- Kagi, D.; Ledermann, B.; Burki, K.; Seiler, P.; Odermatt, B.; Olsen, K.J.; Podack, E.R.; Zinkernagel, R.M.; Hengartner, H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994, 369, 31–37. [Google Scholar] [CrossRef]
- Murali-Krishna, K.; Altman, J.D.; Suresh, M.; Sourdive, D.J.D.; Zajac, A.J.; Miller, J.D.; Slansky, J.; Ahmed, R. Counting antigen-specific CD8 T cells: A reevaluation of bystander activation during viral infection. Immunity 1998, 8, 177–187. [Google Scholar] [CrossRef]
- Miller, J.D.; van der Most, R.G.; Akondy, R.S.; Glidewell, J.T.; Albott, S.; Masopust, D.; Murali-Krishna, K.; Mahar, P.L.; Edupuganti, S.; Lalor, S.; et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008, 28, 710–722. [Google Scholar] [CrossRef]
- Lau, L.L.; Jamieson, B.D.; Somasundaram, T.; Ahmed, R. Cytotoxic T-cell memory without antigen. Nature 1994, 369, 648–652. [Google Scholar] [CrossRef]
- Murali-Krishna, K.; Lau, L.L.; Sambhara, S.; Lemonnier, F.; Altman, J.; Ahmed, R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science 1999, 286, 1377–1381. [Google Scholar] [CrossRef]
- Blattman, J.N.; Antia, R.; Sourdive, D.J.; Wang, X.; Kaech, S.M.; Murali-Krishna, K.; Altman, J.D.; Ahmed, R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J. Exp. Med. 2002, 195, 657–664. [Google Scholar] [CrossRef]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar]
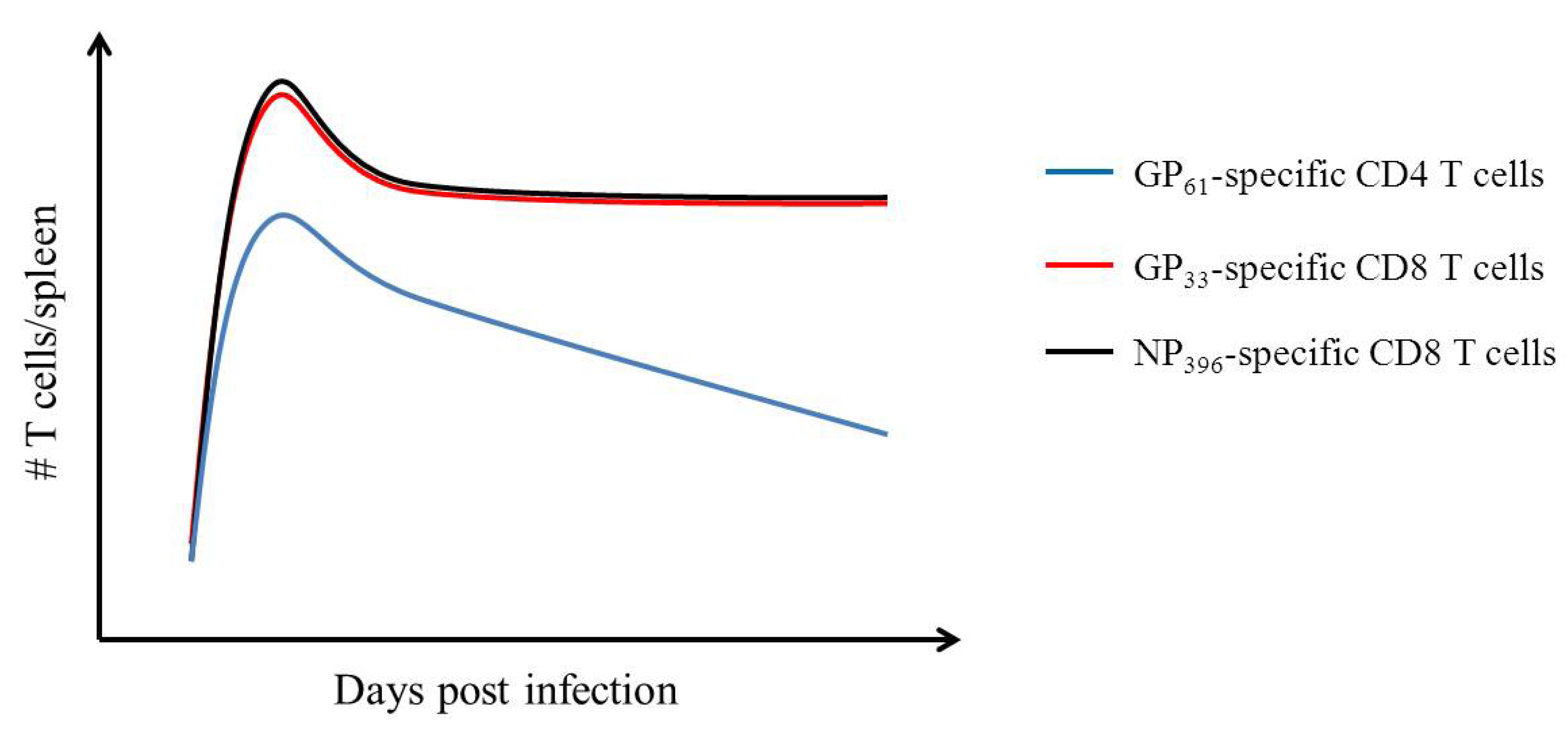
- Homann, D.; Teyton, L.; Oldstone, M.B. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 2001, 7, 913–919. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Wherry, E.J.; Teichgraber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar]
- Baron, V.; Bouneaud, C.; Cumano, A.; Lim, A.; Arstila, T.P.; Kourilsky, P.; Ferradini, L.; Pannetier, C. The repertoires of circulating human CD8(+) central and effector memory T cell subsets are largely distinct. Immunity 2003, 18, 193–204. [Google Scholar] [CrossRef]
- Evans, R.; Alexander, P. Mechanism of immunologically specific killing of tumour cells by macrophages. Nature 1972, 236, 168–170. [Google Scholar]
- Welsh, R.M., Jr.; Zinkernagel, R.M. Heterospecific cytotoxic cell activity induced during the first three days of acute lymphocytic choriomeningitis virus infection in mice. Nature 1977, 268, 646–648. [Google Scholar] [CrossRef]
- Welsh, R.M., Jr. Cytotoxic cells induced during lymphocytic choriomeningitis virus infection of mice. I. Characterization of natural killer cell induction. J. Exp. Med. 1978, 148, 163–181. [Google Scholar] [CrossRef]
- Waggoner, S.N.; Cornberg, M.; Selin, L.K.; Welsh, R.M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2012, 481, 394–398. [Google Scholar]
- Ochsenbein, A.F.; Fehr, T.; Lutz, C.; Suter, M.; Brombacher, F.; Hengartner, H.; Zinkernagel, R.M. Control of early viral and bacterial distribution and disease by natural antibodies. Science 1999, 286, 2156–2159. [Google Scholar] [CrossRef]
- Karrer, U.; Althage, A.; Odermatt, B.; Roberts, C.W.; Korsmeyer, S.J.; Miyawaki, S.; Hengartner, H.; Zinkernagel, R.M. On the key role of secondary lymphoid organs in antiviral immune responses studied in alymphoplastic (aly/aly) and spleenless (Hox11(-)/-) mutant mice. J. Exp. Med. 1997, 185, 2157–2170. [Google Scholar]
- McChesney, M.B.; Oldstone, M.B. Virus-induced immunosuppression: Infections with measles virus and human immunodeficiency virus. Adv. Immunol. 1989, 45, 335–380. [Google Scholar] [CrossRef]
- Mims, C.A.; Wainwright, S. The immunodepressive action of lymphocytic choriomeningitis virus in mice. J. Immunol. 1968, 101, 717–724. [Google Scholar]
- McChesney, M.B.; Oldstone, M.B. Viruses perturb lymphocyte functions: Selected principles characterizing virus-induced immunosuppression. Annu. Rev. Immunol. 1987, 5, 279–304. [Google Scholar] [CrossRef]
- Thomsen, A.R.; Bro-Jorgensen, K.; Jensen, B.L. Lymphocytic choriomeningitis virus-induced immunosuppression: Evidence for viral interference with T-cell maturation. Infect. Immun. 1982, 37, 981–986. [Google Scholar]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.D.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated t cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8623–8628. [Google Scholar] [CrossRef]
- Khaitan, A.; Unutmaz, D. Revisiting immune exhaustion during HIV infection. Curr. HIV/AIDS Rep. 2011, 8, 4–11. [Google Scholar] [CrossRef]
- Kim, P.S.; Ahmed, R. Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol. 2010, 22, 223–230. [Google Scholar]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Pellegrini, M.; Calzascia, T.; Toe, J.G.; Preston, S.P.; Lin, A.E.; Elford, A.R.; Shahinian, A.; Lang, P.A.; Lang, K.S.; Morre, M.; et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 2011, 144, 601–613. [Google Scholar] [CrossRef]
- Nanjappa, S.G.; Kim, E.H.; Suresh, M. Immunotherapeutic effects of IL-7 during a chronic viral infection in mice. Blood 2011, 117, 5123–5132. [Google Scholar]
- Cole, G.A.; Nathanson, N.; Prendergast, R.A. Requirement for theta-bearing cells in lymphocytic choriomeningitis virus-induced central nervous system disease. Nature 1972, 238, 335–337. [Google Scholar]
- Gilden, D.H.; Cole, G.A.; Nathanson, N. Immunopathogenesis of acute central nervous system disease produced by lymphocytic choriomeningitis virus. II. Adoptive immunization of virus carriers. J. Exp. Med. 1972, 135, 874–889. [Google Scholar] [CrossRef]
- Gilden, D.H.; Cole, G.A.; Monjan, A.A.; Nathanson, N. Immunopathogenesis of acute central nervous system disease produced by lymphocytic choriomeningitis virus. I. Cyclophosphamide-mediated induction by the virus-carrier state in adult mice. J. Exp. Med. 1972, 135, 860–873. [Google Scholar] [CrossRef]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 2009, 457, 191–195. [Google Scholar]
- Gresser, I.; Tovey, M.G.; Bandu, M.E.; Maury, C.; Brouty-Boye, D. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum. I. Rapid evolution of encephalomyocarditis virus infection. J. Exp. Med. 1976, 144, 1305–1315. [Google Scholar] [CrossRef]
- Gresser, I.; Tovey, M.G.; Maury, C.; Bandu, M.T. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum. II. Studies with herpes simplex, Moloney sarcoma, vesicular stomatitis, Newcastle disease, and influenza viruses. J. Exp. Med. 1976, 144, 1316–1323. [Google Scholar] [CrossRef]
- Gresser, I.; Maury, C.; Tovey, M.; Morel-Maroger, L.; Pontillon, F. Progressive glomerulonephritis in mice treated with interferon preparations at birth. Nature 1976, 263, 420–422. [Google Scholar] [CrossRef]
- Gresser, I.; Tovey, M.G.; Maury, C.; Chouroulinkov, I. Lethality of interferon preparations for newborn mice. Nature 1975, 258, 76–78. [Google Scholar]
- Riviere, Y.; Gresser, I.; Guillon, J.C.; Tovey, M.G. Inhibition by anti-interferon serum of lymphocytic choriomeningitis virus disease in suckling mice. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 2135–2139. [Google Scholar] [CrossRef]
- Pfizenmaier, K.; Trostmann, H.; Rollinghoff, M.; Wagner, H. Temporary presence of self-reactive cytotoxic T lymphocytes during murine lymphocytic choriomeningitis. Nature 1975, 258, 238–240. [Google Scholar] [CrossRef]
- Pfizenmaier, K.; Trostmann, H.; Rollinghoff, M.; Wagner, H. Cell-mediated immunity in lumphocytic choriomeningitis. I. The specificity of the cytotoxic T lymphocytes. Z. Immunitatsforsch. Exp. Klin. Immunol. 1976, 151, 224–236. [Google Scholar]
- Ohashi, P.S.; Oehen, S.; Buerki, K.; Pircher, H.; Ohashi, C.T.; Odermatt, B.; Malissen, B.; Zinkernagel, R.M.; Hengartner, H. Ablation of "tolerance" and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 1991, 65, 305–317. [Google Scholar] [CrossRef]
- Oldstone, M.B.; Nerenberg, M.; Southern, P.; Price, J.; Lewicki, H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell 1991, 65, 319–331. [Google Scholar]
- Evans, C.F.; Horwitz, M.S.; Hobbs, M.V.; Oldstone, M.B. Viral infection of transgenic mice expressing a viral protein in oligodendrocytes leads to chronic central nervous system autoimmune disease. J. Exp. Med. 1996, 184, 2371–2384. [Google Scholar] [CrossRef]
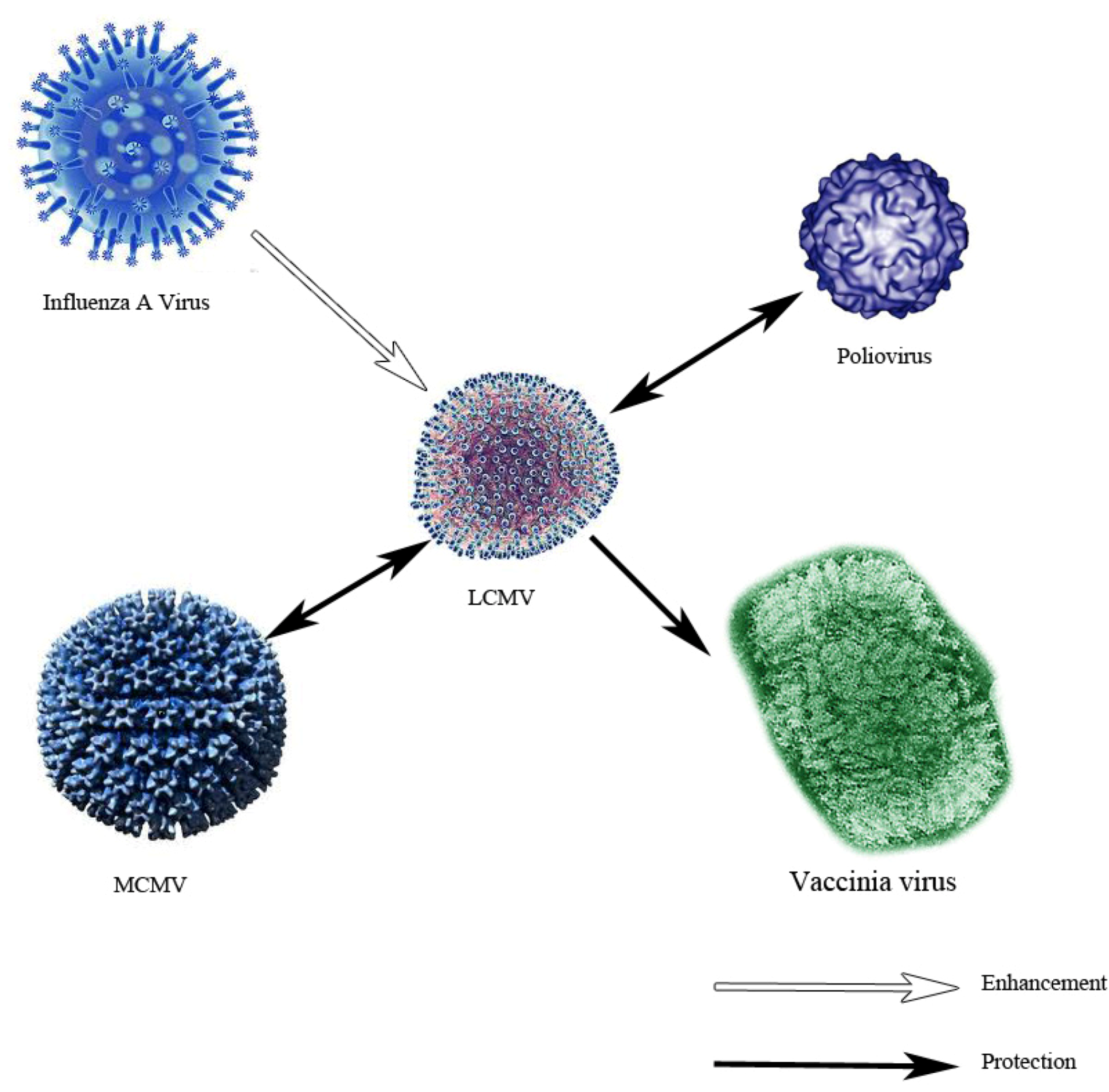
- Cornberg, M.; Sheridan, B.S.; Saccoccio, F.M.; Brehm, M.A.; Selin, L.K. Protection against vaccinia virus challenge by CD8 memory T cells resolved by molecular mimicry. J. Virol. 2007, 81, 934–944. [Google Scholar] [CrossRef]
- Chen, H.D.; Fraire, A.E.; Joris, I.; Brehm, M.A.; Welsh, R.M.; Selin, L.K. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat. Immunol. 2001, 2, 1067–1076. [Google Scholar]
- Welsh, R.M.; Che, J.W.; Brehm, M.A.; Selin, L.K. Heterologous immunity between viruses. Immunol. Rev. 2010, 235, 244–266. [Google Scholar]
- Selin, L.K.; Nahill, S.R.; Welsh, R.M. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J. Exp. Med. 1994, 179, 1933–1943. [Google Scholar] [CrossRef]
- Bergthaler, A.; Flatz, L.; Hegazy, A.N.; Johnson, S.; Horvath, E.; Lohning, M.; Pinschewer, D.D. Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 21641–21646. [Google Scholar]
- Sullivan, B.M.; Emonet, S.F.; Welch, M.J.; Lee, A.M.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 2969–2974. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009, 458, 206–210. [Google Scholar]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.-R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Marshall, H.D.; Chandele, A.; Jung, Y.W.; Meng, H.; Poholek, A.C.; Parish, I.A.; Rutishauser, R.; Cui, W.; Kleinstein, S.H.; Craft, J.; et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 2011, 35, 633–646. [Google Scholar] [CrossRef]
- Amanna, I.J.; Raue, H.P.; Slifka, M.K. Development of a new hydrogen peroxide-based vaccine platform. Nat. Med. 2012, 18, 974–979. [Google Scholar]
- Flatz, L.; Cheng, C.; Wang, L.; Foulds, K.; Ko, S.Y.; Kong, W.P.; Roychoudhuri, R.; Shi, W.; Bao, S.; Todd, J.P.; et al. Gene-based vaccination with a mis-matched envelope protects against simian immunodeficiency virus infection in non-human primates. J. Virol. 2012, 86, 7760–7770. [Google Scholar]
- Flatz, L.; Hegazy, A.N.; Bergthaler, A.; Verschoor, A.; Claus, C.; Fernandez, M.; Gattinoni, L.; Johnson, S.; Kreppel, F.; Kochanek, S.; et al. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat. Med. 2010, 16, 339–345. [Google Scholar] [CrossRef]
- Folk, S.; Steinbecker, S.; Windmeyer, J.; Macneil, A.; Campbell, S.; Rollin, P.E. Lymphocytic choriomeningitis with severe manifestations, Missouri, USA. Emerg. Infect. Dis. 2011, 17, 1973–1974. [Google Scholar] [CrossRef]
- Razonable, R.R. Rare, unusual, and less common virus infections after organ transplantation. Curr. Opin. Organ. Transplant. 2011, 16, 580–587. [Google Scholar] [CrossRef]
- Stahl, J.P.; Mailles, A.; Dacheux, L.; Morand, P. Epidemiology of viral encephalitis in 2011. Med. Mal. Infect. 2011, 41, 453–464. [Google Scholar]
- Milazzo, M.L.; Campbell, G.L.; Fulhorst, C.F. Novel arenavirus infection in humans, United States. Emerg. Infect. Dis. 2011, 17, 1417–1420. [Google Scholar]
- Grant-Klein, R.J.; Altamura, L.A.; Schmaljohn, C.S. Progress in recombinant DNA-derived vaccines for Lassa virus and filoviruses. Virus Res. 2011, 162, 148–161. [Google Scholar] [CrossRef]
- Lan, S.; McLay Schelde, L.; Wang, J.; Kumar, N.; Ly, H.; Liang, Y. Development of infectious clones for virulent and avirulent pichinde viruses: A model virus to study arenavirus-induced hemorrhagic fevers. J. Virol. 2009, 83, 6357–6362. [Google Scholar]
- Albarino, C.G.; Bergeron, E.; Erickson, B.R.; Khristova, M.L.; Rollin, P.E.; Nichol, S.T. Efficient reverse genetics generation of infectious junin viruses differing in glycoprotein processing. J. Virol. 2009, 83, 5606–5614. [Google Scholar] [CrossRef]
- Flatz, L.; Bergthaler, A.; de la Torre, J.C.; Pinschewer, D.D. Recovery of an arenavirus entirely from RNA polymerase I/II-driven cDNA. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4663–4668. [Google Scholar]
- Popkin, D.L.; Teijaro, J.R.; Sullivan, B.M.; Urata, S.; Rutschmann, S.; de la Torre, J.C.; Kunz, S.; Beutler, B.; Oldstone, M. Hypomorphic mutation in the site-1 protease Mbtps1 endows resistance to persistent viral infection in a cell-specific manner. Cell Host Microbe 2011, 9, 212–222. [Google Scholar] [CrossRef]
- Sanchez, A.B.; de la Torre, J.C. Rescue of the prototypic Arenavirus LCMV entirely from plasmid. Virology 2006, 350, 370–380. [Google Scholar] [CrossRef]
- Popkin, D.L.; Teijaro, J.R.; Lee, A.M.; Lewicki, H.; Emonet, S.; de la Torre, J.C.; Oldstone, M. Expanded potential for recombinant trisegmented lymphocytic choriomeningitis viruses: Protein production, antibody production, and in vivo assessment of biological function of genes of interest. J. Virol. 2011, 85, 7928–7932. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, X.; Ramachandran, S.; Mann, M.; Popkin, D.L. Role of Lymphocytic Choriomeningitis Virus (LCMV) in Understanding Viral Immunology: Past, Present and Future. Viruses 2012, 4, 2650-2669. https://doi.org/10.3390/v4112650
Zhou X, Ramachandran S, Mann M, Popkin DL. Role of Lymphocytic Choriomeningitis Virus (LCMV) in Understanding Viral Immunology: Past, Present and Future. Viruses. 2012; 4(11):2650-2669. https://doi.org/10.3390/v4112650
Chicago/Turabian StyleZhou, Xin, Srividya Ramachandran, Margaret Mann, and Daniel L. Popkin. 2012. "Role of Lymphocytic Choriomeningitis Virus (LCMV) in Understanding Viral Immunology: Past, Present and Future" Viruses 4, no. 11: 2650-2669. https://doi.org/10.3390/v4112650