Identification of Genes Critical for Resistance to Infection by West Nile Virus Using RNA-Seq Analysis


Abstract
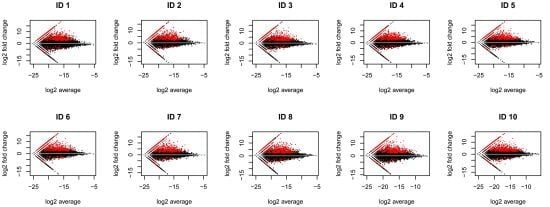
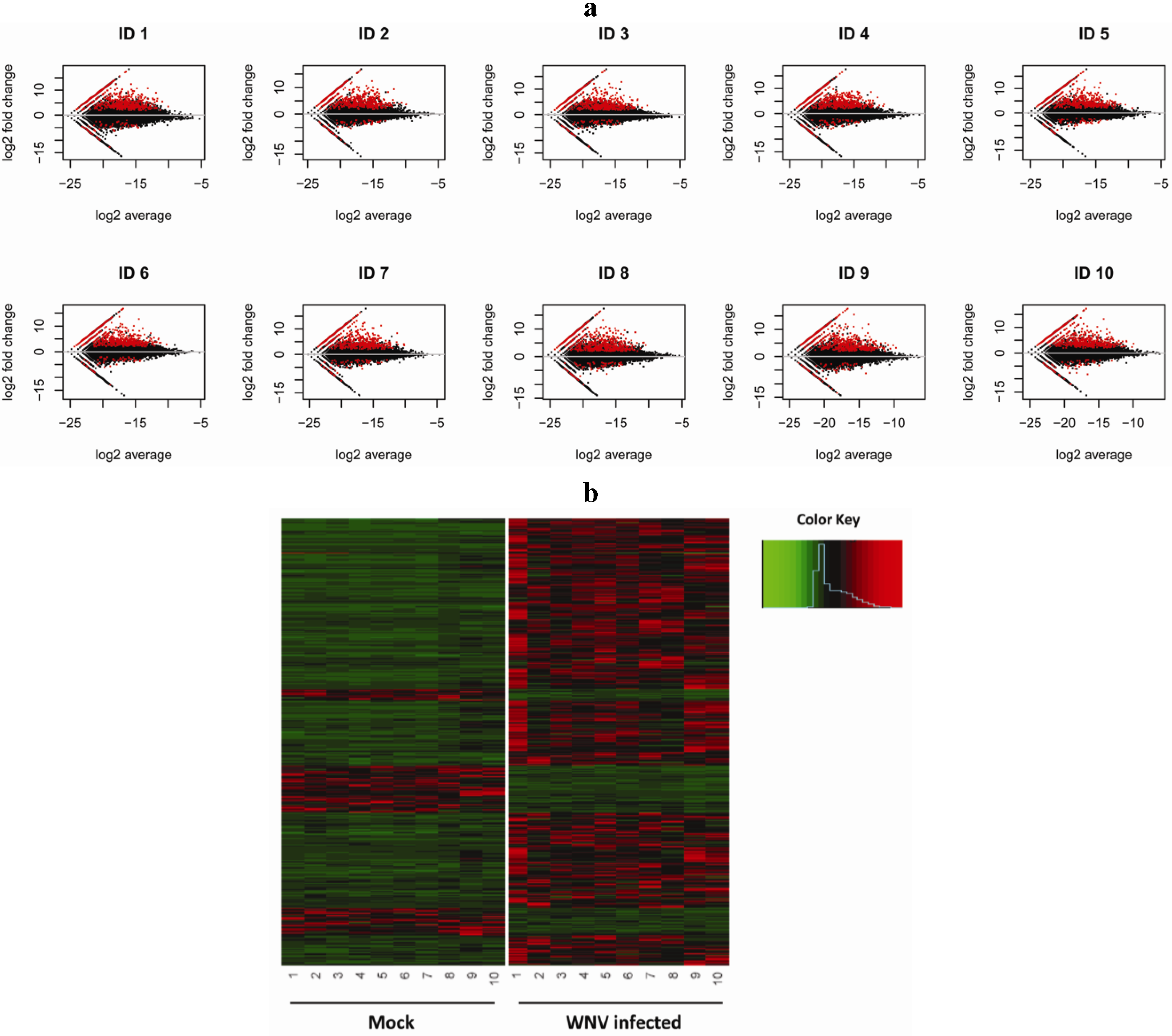
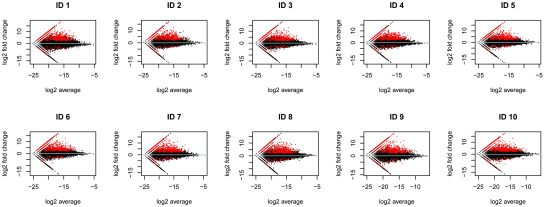
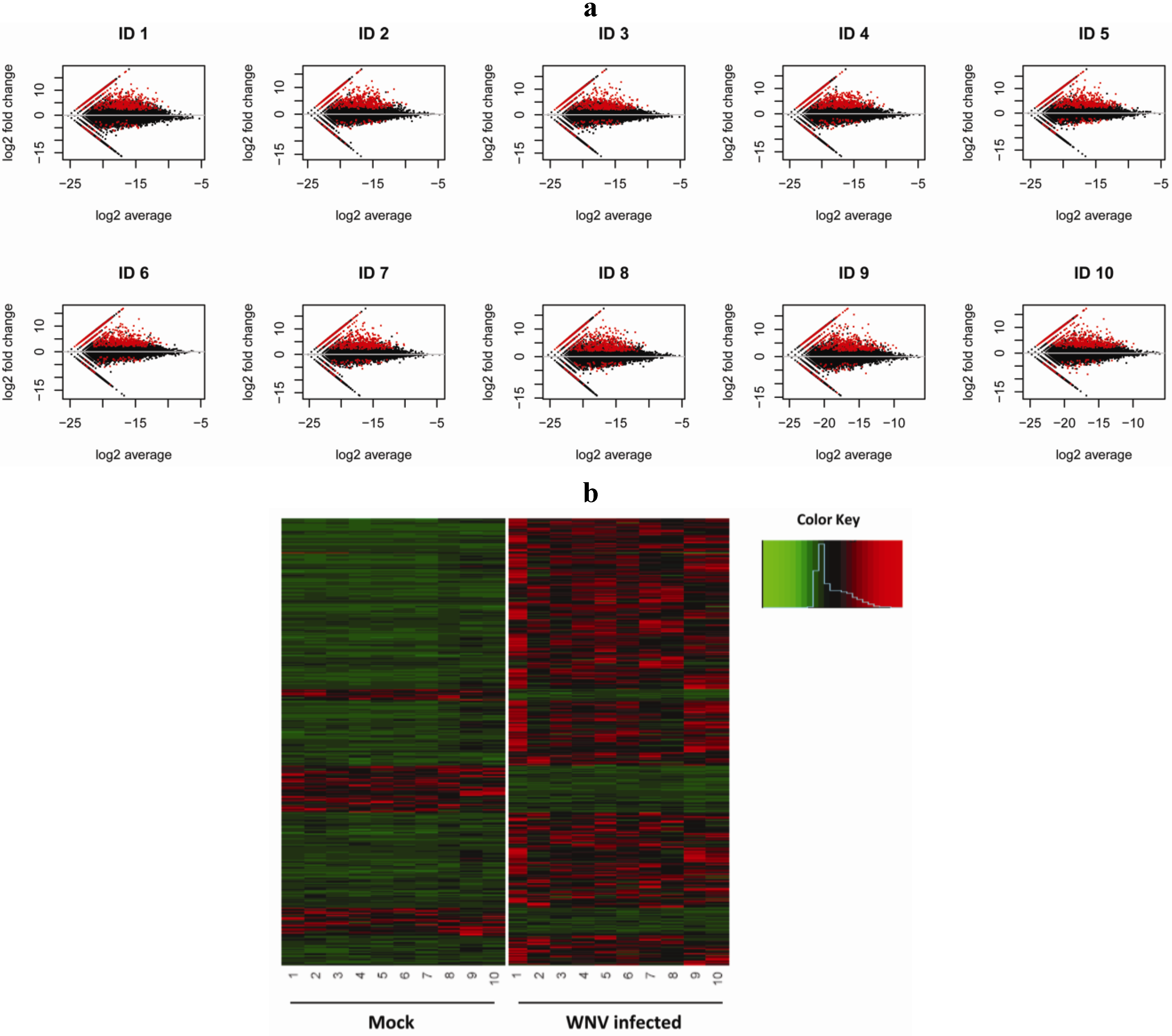
:
1. Introduction
2. Results and Discussion
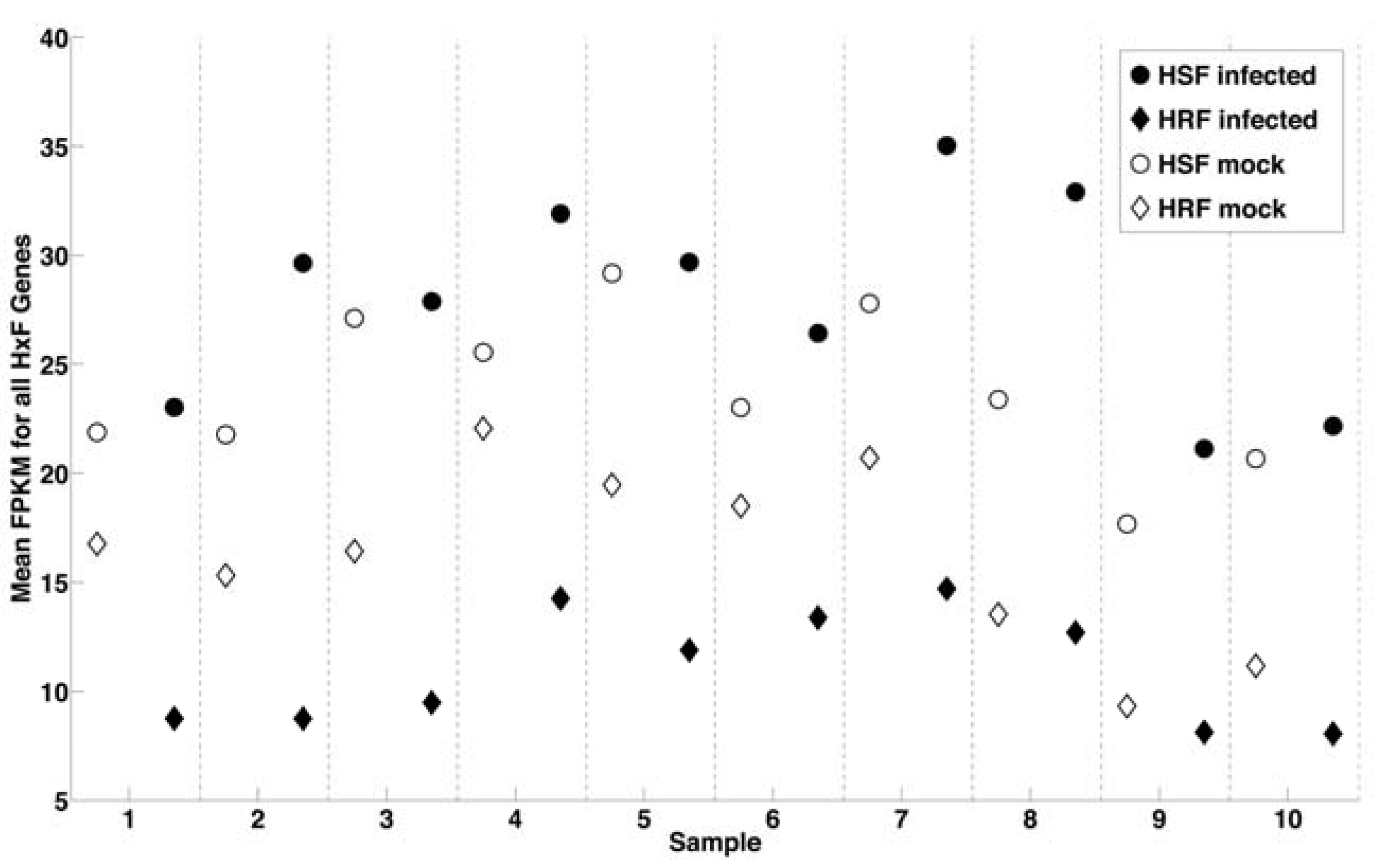
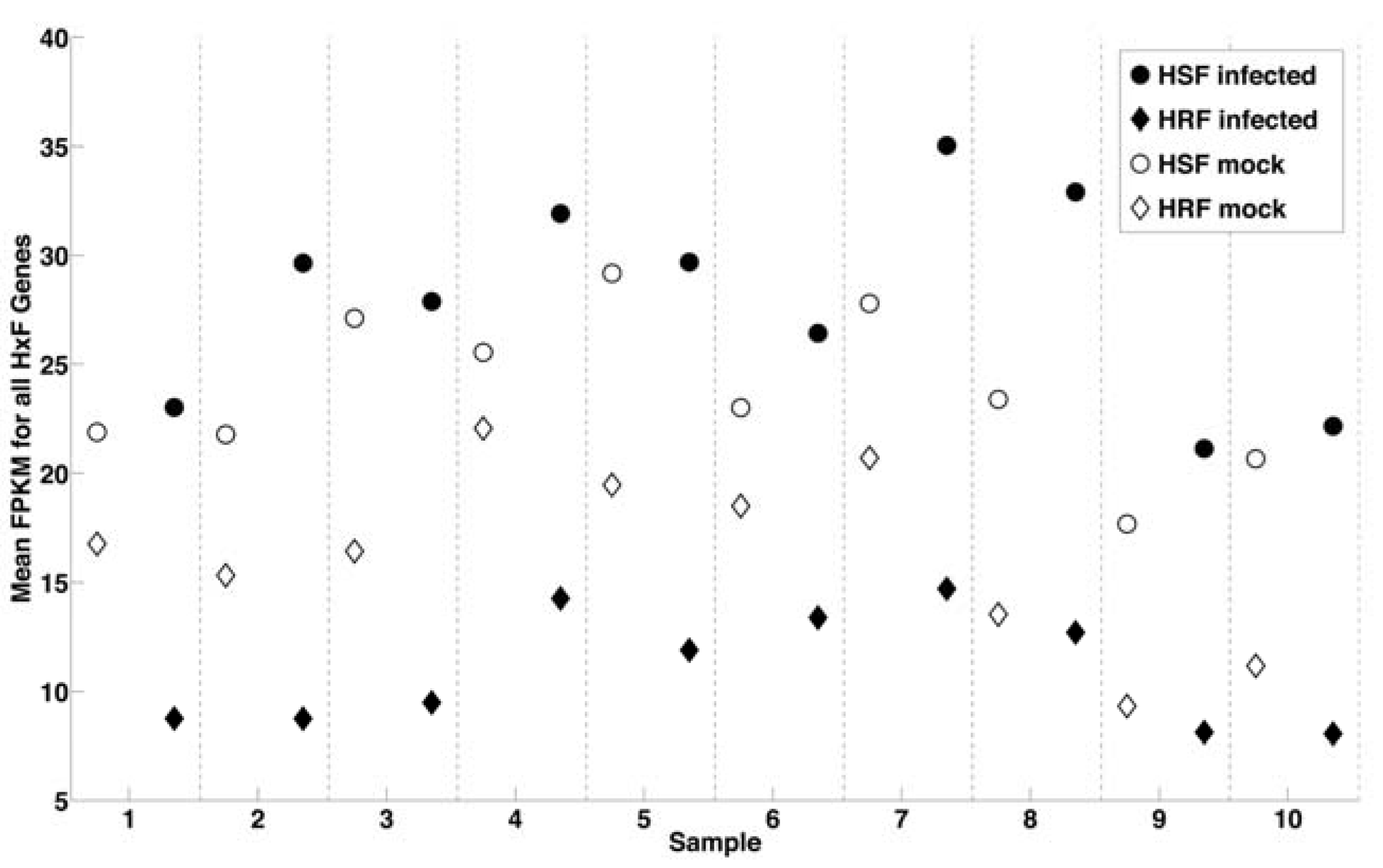
2.1. RNA-Seq Analysis of Differential Gene Expression by Human Macrophages Infected with WNV
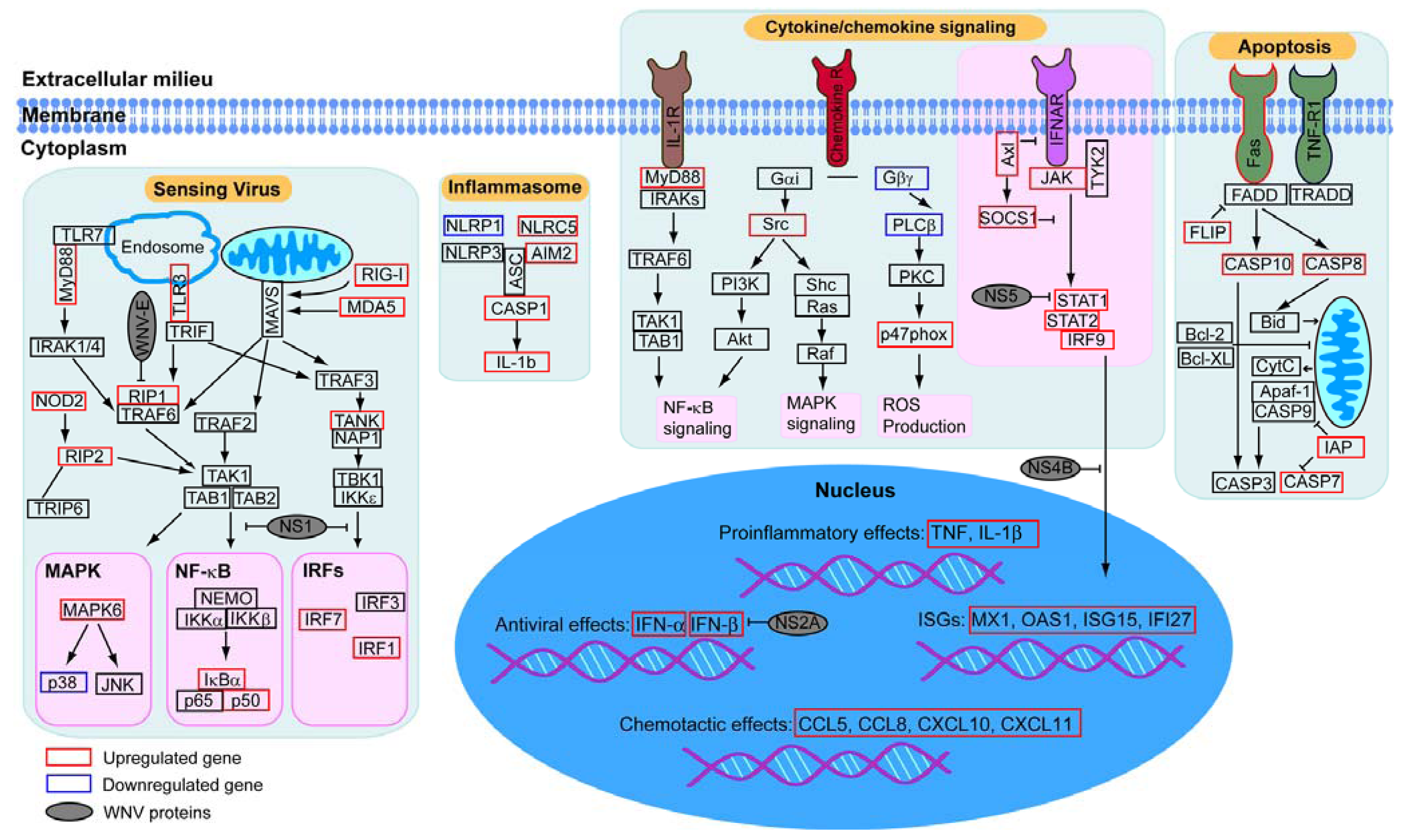
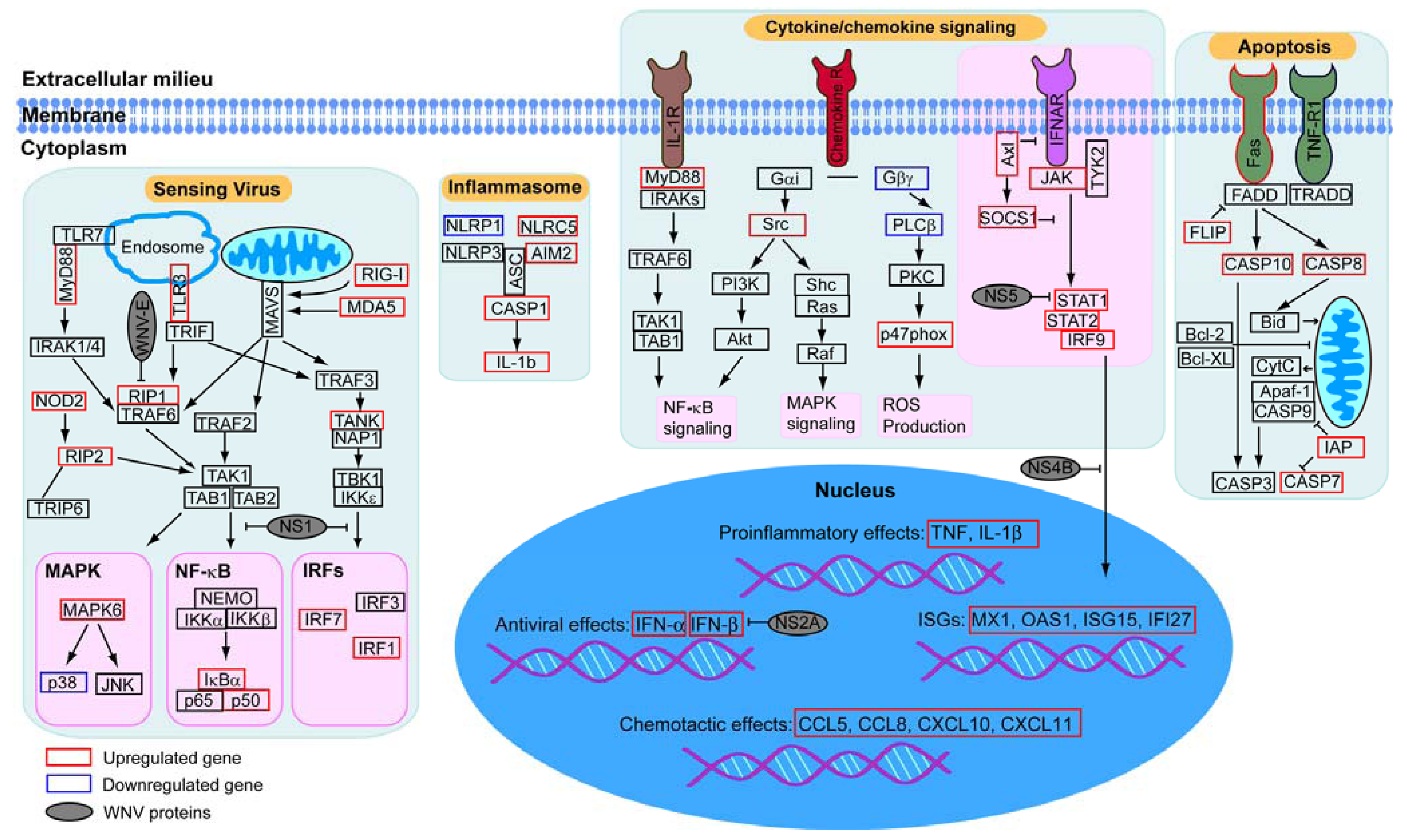
2.2. Virally Induced Pathways Identified by Functional Annotation Clustering

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Count | PValue | FDR |
|---|---|---|---|---|
| Cluster 1 | Enrichment Score: 20.92 | |||
| GO: BP | GO:0006952~defense response | 101 | 2.50 × 10−25 | 4.53 × 10−22 |
| GO: BP | GO:0006954~inflammatory response | 64 | 3.66 × 10−20 | 6.64 × 10−17 |
| GO: BP | GO:0009611~response to wounding | 83 | 1.80 × 10−19 | 3.27 × 10−16 |
| Cluster 2 | Enrichment Score: 9.21 | |||
| GO: BP | GO:0001775~cell activation | 46 | 2.83 × 10−11 | 5.13 × 10−8 |
| GO: BP | GO:0045321~leukocyte activation | 40 | 2.67 × 10−10 | 4.84 × 10−7 |
| GO: BP | GO:0046649~lymphocyte activation | 34 | 3.07 × 10−9 | 5.57 × 10−6 |
| GO: BP | GO:0042110~T cell activation | 26 | 6.22 × 10−9 | 1.13 × 10−5 |
| Cluster 3 | Enrichment Score: 6.62 | |||
| SP_PIR_KEYWORDS | inflammatory response | 26 | 4.45 × 10−15 | 6.43 × 10−12 |
| SP_PIR_KEYWORDS | chemotaxis | 22 | 7.21 × 10−12 | 1.04 × 10−8 |
| GO: MF | GO:0005125~cytokine activity | 37 | 2.14 × 10−11 | 3.31 × 10−8 |
| SP_PIR_KEYWORDS | cytokine | 33 | 7.05 × 10−11 | 1.02 × 10−7 |
| GO: MF | GO:0042379~chemokine receptor binding | 18 | 1.62 × 10−10 | 2.50 × 10−7 |
| INTERPRO | IPR001811:Small chemokine, interleukin-8-like | 16 | 3.86 × 10−10 | 6.24 × 10−7 |
| GO: MF | GO:0008009~chemokine activity | 17 | 5.38 × 10−10 | 8.32 × 10−7 |
| SMART | SM00199:SCY | 16 | 2.96 × 10−9 | 3.89 × 10−6 |
| KEGG_PATHWAY | hsa04060:Cytokine-cytokine receptor interaction | 44 | 4.30 × 10−9 | 5.16 × 10−6 |
| SP_PIR_KEYWORDS | inflammation | 12 | 5.28 × 10−9 | 7.64 × 10−6 |
| GO: BP | GO:0006935~chemotaxis | 27 | 2.20 × 10−7 | 3.99 × 10−4 |
| GO: BP | GO:0042330~taxis | 27 | 2.20 × 10−7 | 3.99 × 10−4 |
| GO: CC | GO:0005615~extracellular space | 65 | 2.68 × 10−7 | 3.73 × 10−4 |
| KEGG_PATHWAY | hsa04062:Chemokine signaling pathway | 32 | 5.92 × 10−7 | 7.10 × 10−4 |
| GO: BP | GO:0007626~locomotory behavior | 35 | 2.35 × 10−6 | 4.26 × 10−3 |
| PIR_SUPERFAMILY | PIRSF001950:small inducible chemokine, C/CC types | 9 | 1.28 × 10−5 | 1.78 × 10−2 |
| INTERPRO | IPR000827:Small chemokine, C-C group, conserved site | 9 | 1.66 × 10−5 | 2.68 × 10−2 |
| Cluster 4 | Enrichment Score: 6.40 | |||
| GO: BP | GO:0001817~regulation of cytokine production | 33 | 1.03 × 10−9 | 1.86 × 10−6 |
| GO: BP | GO:0051240~positive regulation of multicellular organismal process | 32 | 3.97 × 10−6 | 7.21 × 10−3 |
| GO: BP | GO:0001819~positive regulation of cytokine production | 17 | 1.47 × 10−5 | 2.67 × 10−2 |
| Cluster 5 | Enrichment Score: 6.32 | |||
| GO: BP | GO:0002237~response to molecule of bacterial origin | 20 | 6.81 × 10−8 | 1.24 × 10−4 |
| GO: BP | GO:0034097~response to cytokine stimulus | 19 | 9.11 × 10−8 | 1.65 × 10−4 |
| GO: BP | GO:0032496~response to lipopolysaccharide | 18 | 3.37 × 10−7 | 6.11 × 10−4 |
| GO: BP | GO:0009617~response to bacterium | 26 | 2.43 × 10−5 | 4.41 × 10−2 |
| Cluster 6 | Enrichment Score: 5.95 | |||
| GO: BP | GO:0042981~regulation of apoptosis | 87 | 1.03 × 10−10 | 1.86 × 10−7 |
| GO: BP | GO:0043067~regulation of programmed cell death | 87 | 1.65 × 10−10 | 3.00 × 10−7 |
| GO: BP | GO:0010941~regulation of cell death | 87 | 1.99 × 10−10 | 3.61 × 10−7 |
| GO: BP | GO:0043065~positive regulation of apoptosis | 48 | 1.20 × 10−6 | 2.18 × 10−3 |
| GO: BP | GO:0043068~positive regulation of programmed cell death | 48 | 1.46 × 10−6 | 2.65 × 10−3 |
| GO: BP | GO:0010942~positive regulation of cell death | 48 | 1.67 × 10−6 | 3.03 × 10−3 |
| GO: BP | GO:0006916~anti-apoptosis | 27 | 2.66 × 10−5 | 4.82 × 10−2 |
| GO: BP | GO:0006917~induction of apoptosis | 36 | 2.75 × 10−5 | 4.98 × 10−2 |
| Cluster 7 | Enrichment Score: 5.33 | |||
| GO: BP | GO:0002684~positive regulation of immune system process | 42 | 1.05 × 10−11 | 1.91 × 10−8 |
| GO: BP | GO:0050865~regulation of cell activation | 29 | 1.06 × 10−7 | 1.92 × 10−4 |
| GO: BP | GO:0002694~regulation of leukocyte activation | 28 | 1.26 × 10−7 | 2.29 × 10−4 |
| GO: BP | GO:0051249~regulation of lymphocyte activation | 26 | 1.75 × 10−7 | 3.17 × 10−4 |
| GO: BP | GO:0050671~positive regulation of lymphocyte proliferation | 15 | 5.79 × 10−7 | 1.05 × 10−3 |
| GO: BP | GO:0032946~positive regulation of mononuclear cell proliferation | 15 | 7.35 × 10−7 | 1.33 × 10−3 |
| GO: BP | GO:0070665~positive regulation of leukocyte proliferation | 15 | 7.35 × 10−7 | 1.33 × 10−3 |
| GO: BP | GO:0050867~positive regulation of cell activation | 21 | 1.04 × 10−6 | 1.88 × 10−3 |
| GO: BP | GO:0050670~regulation of lymphocyte proliferation | 18 | 1.04 × 10−6 | 1.89 × 10−3 |
| GO: BP | GO:0032944~regulation of mononuclear cell proliferation | 18 | 1.24 × 10−6 | 2.26 × 10−3 |
| GO: BP | GO:0070663~regulation of leukocyte proliferation | 18 | 1.24 × 10−6 | 2.26 × 10−3 |
| GO: BP | GO:0002696~positive regulation of leukocyte activation | 20 | 2.08 × 10−6 | 3.78 × 10−3 |
| GO: BP | GO:0051251~positive regulation of lymphocyte activation | 19 | 2.30 × 10−6 | 4.17 × 10−3 |
| GO: BP | GO:0050863~regulation of T cell activation | 20 | 9.40 × 10−6 | 1.71 × 10−2 |
| Cluster 8 | Enrichment Score: 5.24 | |||
| GO: BP | GO:0043122~regulation of I-kappaB kinase/NF-kappaB cascade | 21 | 5.63 × 10−7 | 1.02 × 10−3 |
| GO: BP | GO:0043123~positive regulation of I-kappaB kinase/NF-kappaB cascade | 19 | 2.30 × 10−6 | 4.17 × 10−3 |
| GO: BP | GO:0010647~positive regulation of cell communication | 39 | 3.50 × 10−6 | 6.36 × 10−3 |
| GO: BP | GO:0010740~positive regulation of protein kinase cascade | 25 | 5.99 × 10−6 | 1.09 × 10−2 |
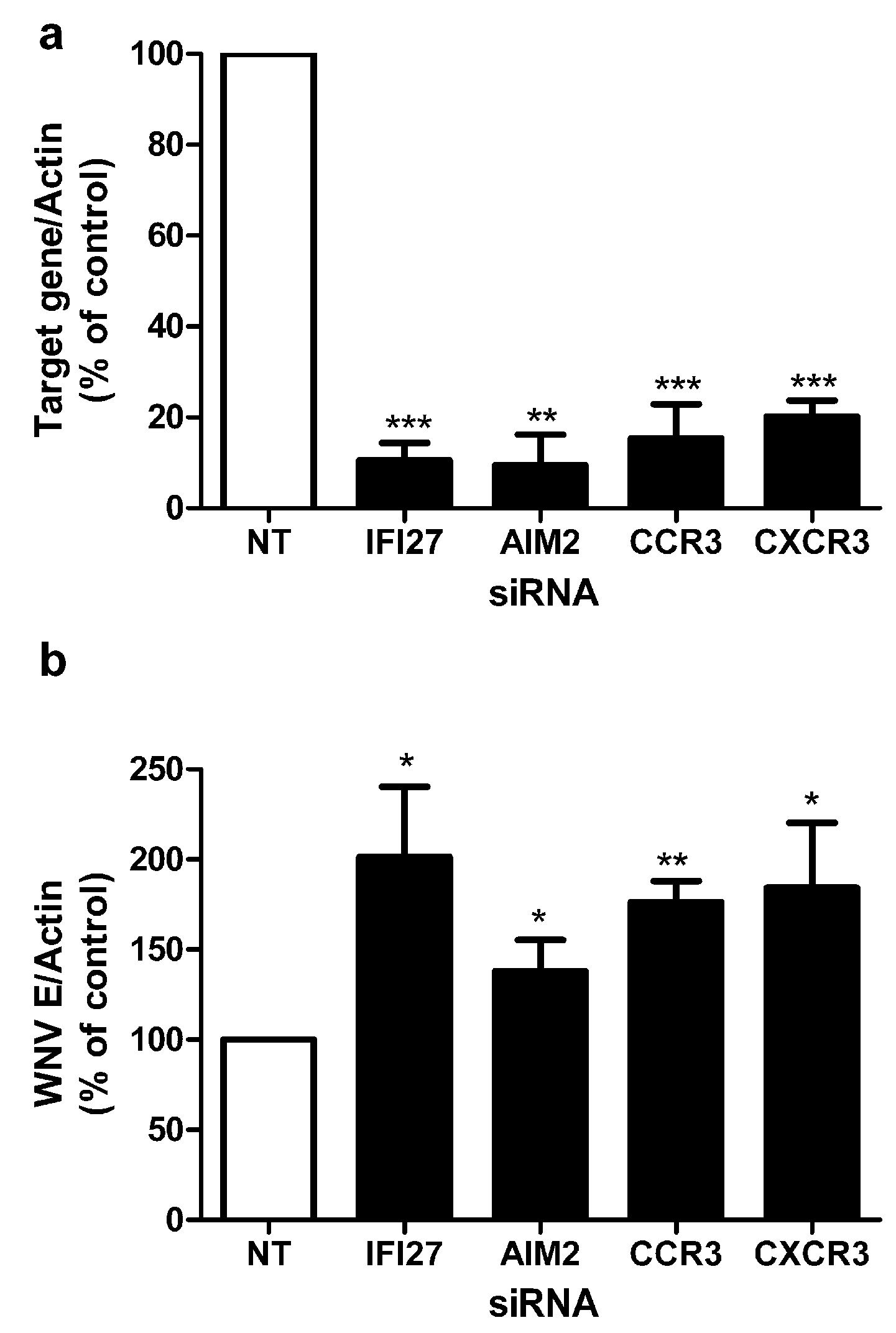
2.3. Correlation of Gene Expression Changes with Genome-Wide RNAi Analysis
2.4. RNAi Knockdown of Novel Genes Shows Critical Role in Resistance to WNV Infection
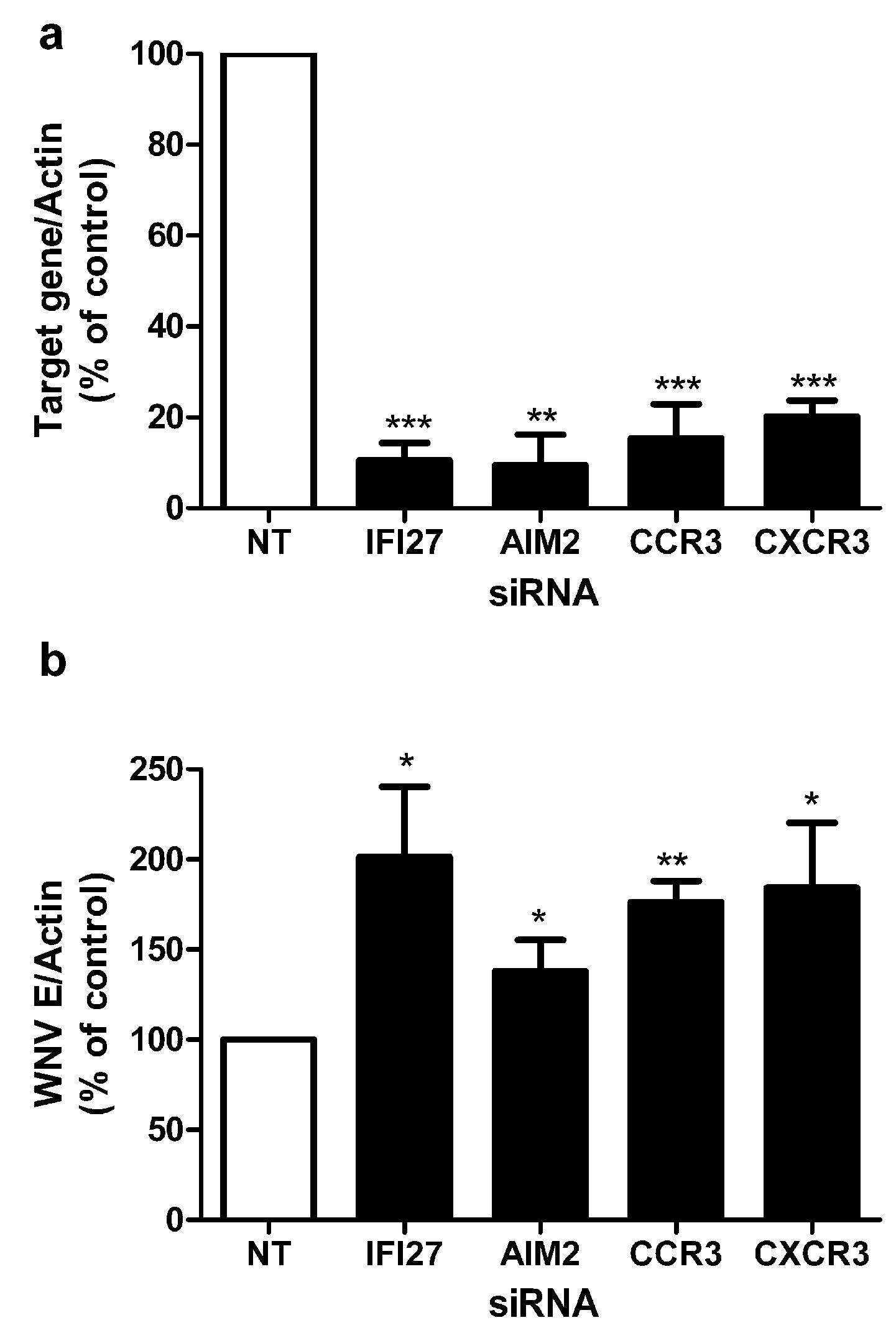
3. Experimental Materials and Methods
3.1. Blood Donors and Preparation of Cells
3.2. WNV Strains and Infections
3.3. RNA Interference
3.4. Quantitative Polymerase Chain Reaction (qPCR)
3.5. Preparation of Libraries for Illumina Deep-Sequencing
3.6. RNA-Seq Analysis
4. Conclusions
Acknowledgements
Conflict of Interest
References and Notes
- Brinton, M.A. The molecular biology of West Nile virus: A new invader of the western hemisphere. Annu. Rev. Microbiol. 2002, 56, 371–402. [Google Scholar] [CrossRef]
- Gubler, D.J. The continuing spread of West Nile virus in the western hemisphere. Clin. Infect. Dis. 2007, 45, 1039–1046. [Google Scholar] [CrossRef]
- Debiasi, R.L.; Tyler, K.L. West Nile virus meningoencephalitis. Nat. Clin. Pract. 2006, 2, 264–275. [Google Scholar] [CrossRef]
- Petersen, L.R.; Carson, P.J.; Biggerstaff, B.J.; Custer, B.; Borchardt, S.M.; Busch, M.P. Estimated cumulative incidence of West Nile virus infection in US adults, 1999–2010. Epidemiol. Infect. 2013, 141, 591–595. [Google Scholar] [CrossRef]
- Lanteri, M.C.; O'Brien, K.M.; Purtha, W.E.; Cameron, M.J.; Lund, J.M.; Owen, R.E.; Heitman, J.W.; Custer, B.; Hirschkorn, D.F.; Tobler, L.H.; et al. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J. Clin. Investig. 2009, 119, 3266–3277. [Google Scholar]
- Wang, T.; Gao, Y.; Scully, E.; Davis, C.T.; Anderson, J.F.; Welte, T.; Ledizet, M.; Koski, R.; Madri, J.A.; Barrett, A.; et al. Gamma delta T cells facilitate adaptive immunity against West Nile virus infection in mice. J. Immunol. 2006, 177, 1825–1832. [Google Scholar]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Conway, M.J.; Montgomery, R.R.; Fikrig, E. West Nile virus: Biology, transmission and human infection. Clin. Microbiol. Rev. 2012, 25, 635–648. [Google Scholar] [CrossRef]
- Ben-Nathan, D.; Huitinga, I.; Lustig, S.; van Rooijen, N.; Kobiler, D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch. Virol. 1996, 141, 459–469. [Google Scholar] [CrossRef]
- Bai, F.; Kong, K.-F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J. Infect. Dis. 2010, 202, 1804–1812. [Google Scholar] [CrossRef]
- Ross, M.E.; Zhou, X.; Song, G.; Shurtleff, S.A.; Girtman, K.; Williams, W.K.; Liu, H.C.; Mahfouz, R.; Raimondi, S.C.; Lenny, N.; et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 2003, 102, 2951–2959. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA Sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef]
- Wilhelm, B.T.; Marguerat, S.; Watt, S.; Schubert, F.; Wood, V.; Goodhead, I.; Penkett, C.J.; Rogers, J.; Bahler, J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 2008, 453, 1239–1243. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef]
- Lister, R.; O'Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Morin, R.D.; Bainbridge, M.; Fejes, A.; Hirst, M.; Krzywinski, M.; Pugh, T.J.; McDonald, H.; Varhol, R.; Jones, S.J.M.; Marra, M.A. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. BioTech 2008, 45, 81–94. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Kong, K.-F.; Delroux, K.; Wang, X.; Qian, F.; Arjona, A.; Malawista, S.E.; Fikrig, E.; Montgomery, R.R. Dysregulation of TLR3 impairs the innate immune response to West Nile virus in the elderly. J. Virol. 2008, 82, 7613–7623. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Klein, R.S.; Diamond, M.S. Immunological headgear: Antiviral immune responses protect against neuroinvasive West Nile virus. Trends Mol. Med. 2008, 14, 286–294. [Google Scholar] [CrossRef]
- Chung, L.M.; Ferguson, J.P.; Zheng, W.; Qian, F.; Bruno, V.M.; Montgomery, R.R.; Zhao, H. Differential expression analysis for paired RNA-seq data. BMC Bioinformatics 2013, 14, 110–124. [Google Scholar] [CrossRef]
- DAVID Bioinformatics Resources 6.7. National Institute of Allergy and Infectious Diseases (NIAID), NIH. Available online: http://david.abcc.ncifcrf.gov/home.jsp (accessed on 11 June 2013).
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Parquet, M.C.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; Defilippis, V.; Fruh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef]
- Smith, J.L.; Grey, F.E.; Uhrlaub, J.L.; Nikolich-Zugich, J.; Hirsch, A.J. West Nile virus induction of the cellular microRNA, Hs_154, contributes to viral-mediated apoptosis through repression of anti-apoptotoic factors. J. Virol. 2012, 86, 5278–5287. [Google Scholar] [CrossRef]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef]
- Zhang, B.; Chan, Y.K.; Lu, B.; Diamond, M.S.; Klein, R.S. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J. Immunol. 2008, 180, 2641–2649. [Google Scholar]
- Panda, A.; Qian, F.; Mohanty, S.; van Duin, D.; Newman, F.K.; Zhang, L.; Chen, S.; Towle, V.; Belshe, R.B.; Fikrig, E.; et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J. Immunol. 2010, 184, 2518–2527. [Google Scholar] [CrossRef]
- Anderson, J.F.; Andreadis, T.G.; Vossbrinck, C.R.; Tirrell, S.; Wakem, E.M.; French, R.A.; Garmendia, A.E.; Van Kruiningen, H.J. Isolation of West Nile virus from mosquitoes, crows, and a Cooper’s hawk in Connecticut. Science 1999, 286, 2331–2333. [Google Scholar] [CrossRef]
- Qian, F.; Wang, X.; Zhang, l.; Lin, A.; Zhao, H.; Fikrig, E.; Montgomery, R.R. Impaired interferon signaling in dendritic cells from older donors infected in vitro with West Nile virus. J. Infect. Dis. 2011, 203, 1415–1424. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus. Available online: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40718/ (accessed on 17 September 2012).
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.M.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.; Pachter, L. Cufflinks, Transcript assembly, differential expression, and differential regulation for RNA-Seq (v 0.9.3). Available online: http://cufflinks.cbcb.umd.edu/ (accessed on 30 November 2010).
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef]
- Brass, A.L.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; Adams, D.J.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and Dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Lim, J.K.; Lisco, A.; McDermott, D.H.; Huynh, L.; Ward, J.M.; Johnson, B.; Johnson, H.; Pape, J.; Foster, G.A.; Krysztof, D.; et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 2009, 5, e1000321. [Google Scholar] [CrossRef]
- Bigham, A.W.; Buckingham, K.J.; Husain, S.; Emond, M.J.; Bofferding, K.M.; Gildersleeve, H.; Rutherford, A.; Astakhova, N.M.; Perelygin, A.A.; Busch, M.P.; et al. Host genetic risk factors for West Nile virus infection and disease progression. PLoS One 2011, 6, e24745. [Google Scholar] [CrossRef]
- Lim, J.K.; Obara, C.J.; Rivollier, A.; Pletnev, A.G.; Kelsall, B.L.; Murphy, P.M. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J. Immunol. 2011, 186, 471–478. [Google Scholar] [CrossRef]
- Glass, W.G.; McDermott, D.H.; Lim, J.K.; Lekhong, S.; Yu, S.F.; Frank, W.A.; Pape, J.; Cheshier, R.C.; Murphy, P.M. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006, 203, 35–40. [Google Scholar] [CrossRef]
- Lim, J.K.; Louie, C.Y.; Glaser, C.; Jean, C.; Johnson, B.; Johnson, H.; McDermott, D.H.; Murphy, P.M. Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J. Infect. Dis. 2008, 197, 262–265. [Google Scholar] [CrossRef]
- Rosebeck, S.; Leaman, D.W. Mitochondrial localization and pro-apoptotic effects of the interferon-inducible protein ISG12a. Apoptosis 2008, 13, 562–572. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Kumar, M.; Roe, K.; Orillo, B.; Muruve, D.A.; Nerurkar, V.R.; Gale, M., Jr.; Verma, S. Inflammasome adaptor protein apoptosis-associated speck-like protein containig CARD (ASC) is critical for the immune response and survival in West Nile virus encephalitis. J. Virol. 2013, 87, 3655–3667. [Google Scholar] [CrossRef]
- Arjona, A.; Wang, P.; Montgomery, R.R.; Fikrig, E. Innate immune control of West Nile virus infection. Cell. Microbiol. 2011, 13, 1648–1658. [Google Scholar] [CrossRef]
- Shirato, K.; Kimura, T.; Mizutani, T.; Kariwa, H.; Takashima, I. Different chemokine expression in lethal and non-lethal murine West Nile virus infection. J. Med. Virol. 2004, 74, 507–513. [Google Scholar] [CrossRef]
- Daffis, S.; Suthar, M.S.; Szretter, K.J.; Gale, M., Jr.; Diamond, M.S. Induction of IFN-beta and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog. 2009, 5, e1000607. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Hay, S.; Kannourakis, G. A time to kill: Viral manipulation of the cell death program. J. Gen. Virol. 2002, 83, 1547–1564. [Google Scholar]
- Kobayashi, S.; Orba, Y.; Yamaguchi, H.; Kimura, T.; Sawa, H. Accumulation of ubiquitinated proteins is related to West Nile virus-induced neuronal apoptosis. Neuropathology 2012, 32, 398–405. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qian, F.; Chung, L.; Zheng, W.; Bruno, V.; Alexander, R.P.; Wang, Z.; Wang, X.; Kurscheid, S.; Zhao, H.; Fikrig, E.; et al. Identification of Genes Critical for Resistance to Infection by West Nile Virus Using RNA-Seq Analysis. Viruses 2013, 5, 1664-1681. https://doi.org/10.3390/v5071664
Qian F, Chung L, Zheng W, Bruno V, Alexander RP, Wang Z, Wang X, Kurscheid S, Zhao H, Fikrig E, et al. Identification of Genes Critical for Resistance to Infection by West Nile Virus Using RNA-Seq Analysis. Viruses. 2013; 5(7):1664-1681. https://doi.org/10.3390/v5071664
Chicago/Turabian StyleQian, Feng, Lisa Chung, Wei Zheng, Vincent Bruno, Roger P. Alexander, Zhong Wang, Xiaomei Wang, Sebastian Kurscheid, Hongyu Zhao, Erol Fikrig, and et al. 2013. "Identification of Genes Critical for Resistance to Infection by West Nile Virus Using RNA-Seq Analysis" Viruses 5, no. 7: 1664-1681. https://doi.org/10.3390/v5071664
APA StyleQian, F., Chung, L., Zheng, W., Bruno, V., Alexander, R. P., Wang, Z., Wang, X., Kurscheid, S., Zhao, H., Fikrig, E., Gerstein, M., Snyder, M., & Montgomery, R. R. (2013). Identification of Genes Critical for Resistance to Infection by West Nile Virus Using RNA-Seq Analysis. Viruses, 5(7), 1664-1681. https://doi.org/10.3390/v5071664