Feline Foamy Virus-Based Vectors: Advantages of an Authentic Animal Model

Abstract
:1. Introduction

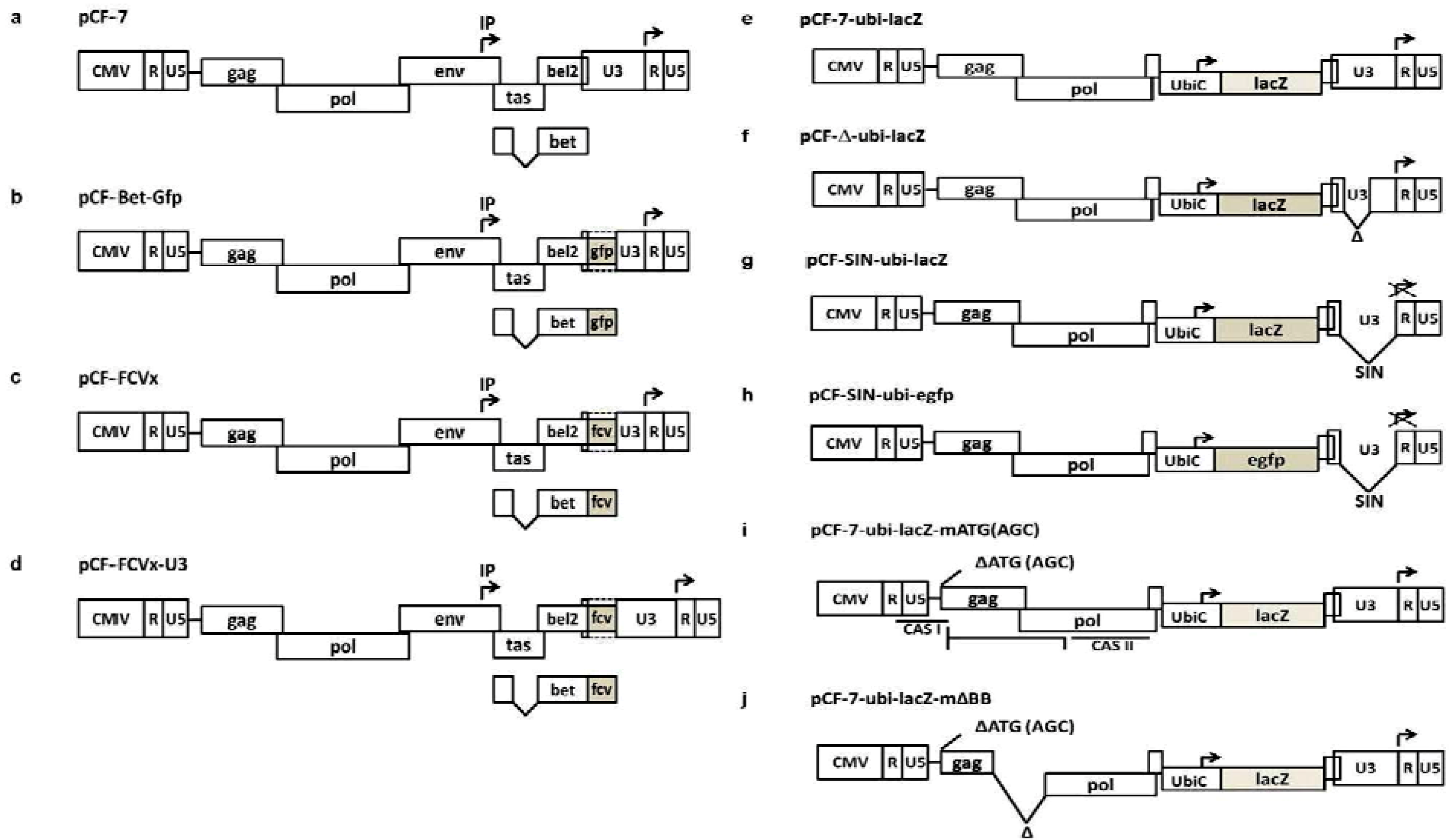{kind=link}
{kind=link}
| Vector | Virus species | Reference | Characteristics |
|---|---|---|---|
| SKY4.0 | FFV/HFV | [23] | Hybrid FFV genome with PFV env and bel1/tas replacing the corresponding FFV sequences |
| pChatul-2/3 | FFV | [26] | wt FFV-F17 genomic vector driven by the CMV-IE promoter instead of the FFV U3 promoter |
| pCF-7 | FFV | [22] | wt FFV-FUV genomic vector driven by the CMV-IE promoter instead of the FFV U3 promoter |
| pCF-Bet-Gfp | FFV | [22] | Gfp marker gene expressed as a Bet-Gfp fusion protein or via an IRES element |
| pCF-FCVx | FFV/FCV | [4] | Recombinant FFV vaccine vectors expressing FCV capsid epitopes as Bet-FCV-E fusion proteins |
2. Construction of Full-Length and Replication-Competent FFV Genomes
3. Replication-Competent FFV Gene Transfer Vectors
4. Replication-Competent FFV Vaccine Vectors
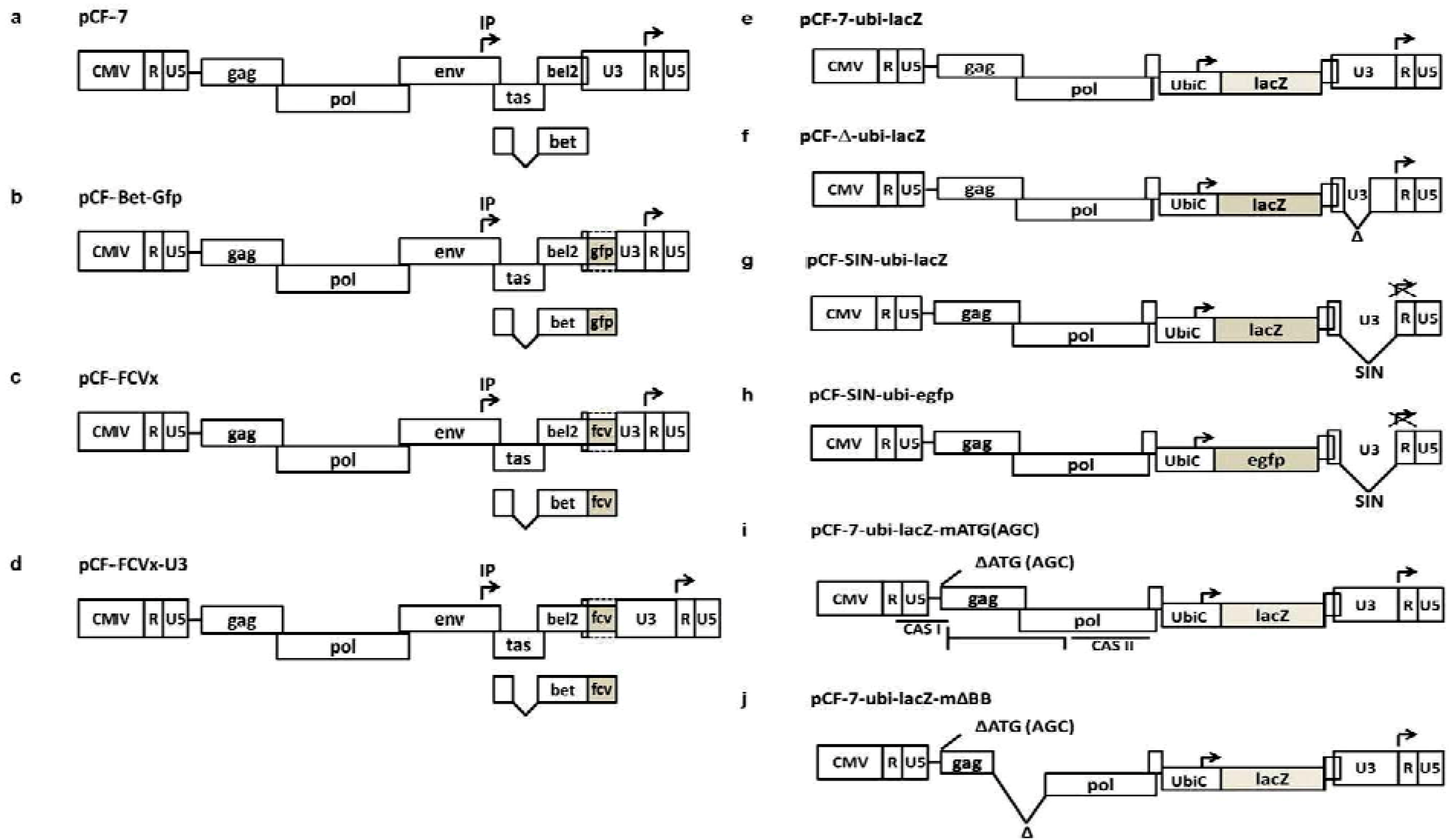
5. Replication-Deficient FFV Vectors
| Vector | Reference | Description |
|---|---|---|
| pCF-SIN pCF-Bet-Gfp-SIN | [58] | Self-inactivating (SIN) FFV Bet-Gfp vectors |
| pCF-7-ubi-lacZ pCF-Δ-ubi-lacZ pCF-SIN-ubi-lacZ | [30] | Env- and bel-deficient FFV vectors expressing the lacZ gene from the ubiC promoter and with intact, truncated or SIN LTR promoters |
| pCF-7-ubi-lacZ-mATG pCF-7-ubi-lacZ-mΔBB | [54] | pCF−/−ubi-lacZ-derived vectors with inactivated gag ATG start codon and gag-pol deletions |
| pCF-SIN-ubi-egfp | described here | pCF-SIN-ubi-lacZ-derived vectors in which the detrimental lacZ reporter gene was replaced by the neutral egfp gene |
5.1. First-Generation RD FFV Vectors
5.2. Second-Generation RD FFV Vectors
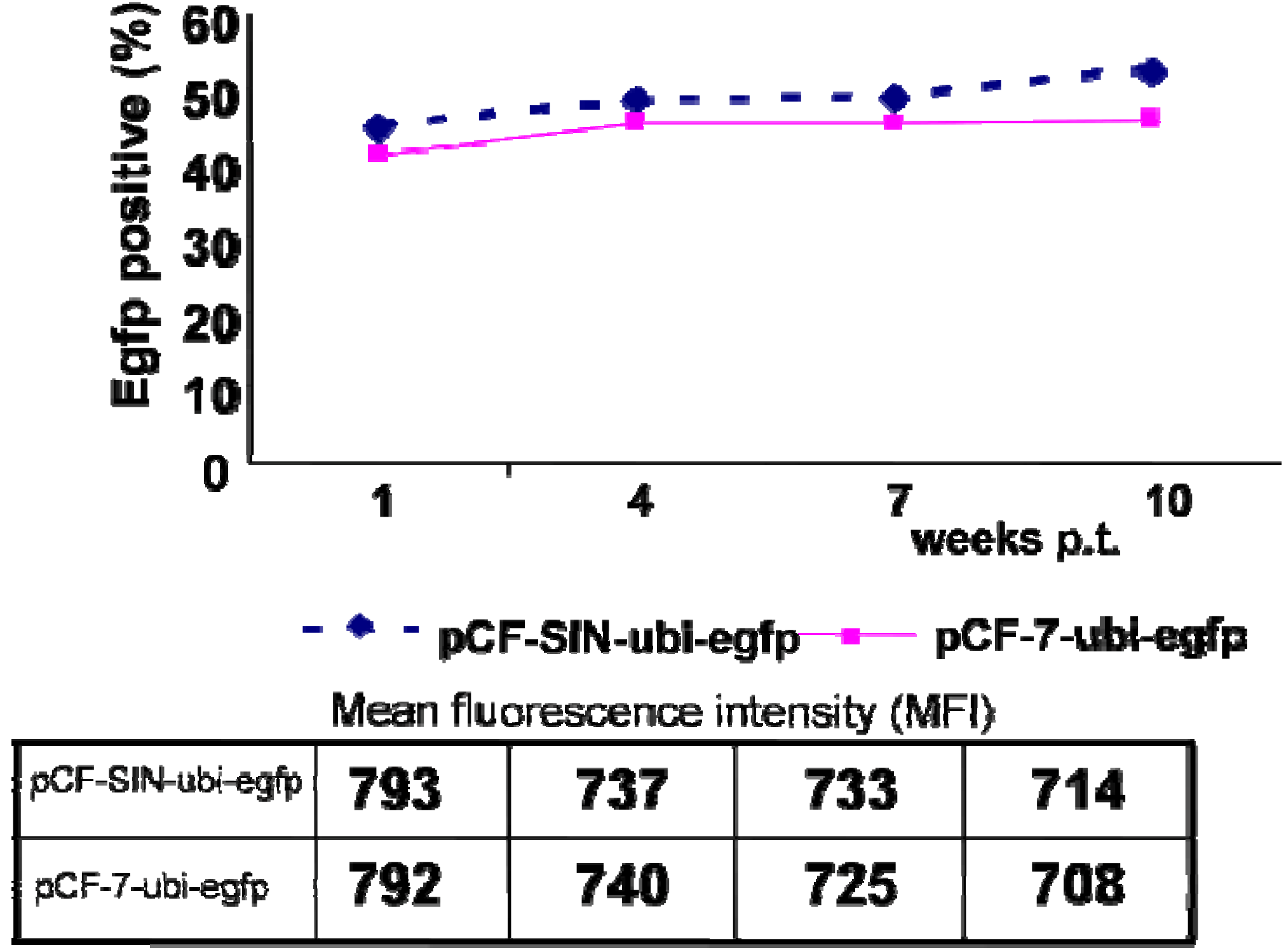
5.3. Third-Generation Gutless FFV Vectors
6. Future Developments
Acknowledgments
Conflict of Interest
References and Notes
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Olson, E.M.; Huo, Y.; Tuschong, L.M.; Allen, J.M.; Li, Y.; Burkholder, T.H.; Russell, D.W. Treatment of canine leukocyte adhesion deficiency by foamy virus vectors expressing CD18 from a PGK promoter. Gene Ther. 2011, 18, 553–559. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Tuschong, L.M.; Calvo, K.R.; Shive, H.R.; Burkholder, T.H.; Karlsson, E.K.; West, R.R.; Russell, D.W.; Hickstein, D.D. Long-term follow-up of foamy viral vector-mediated gene therapy for canine leukocyte adhesion deficiency. Mol. Ther. 2013, 21, 964–972. [Google Scholar] [CrossRef]
- Schwantes, A.; Truyen, U.; Weikel, J.; Weiss, C.; Löchelt, M. Application of chimeric feline foamy virus-based retroviral vectors for the induction of antiviral immunity in cats. J. Virol. 2003, 77, 7830–7842. [Google Scholar] [CrossRef]
- Nowrouzi, A.; Dittrich, M.; Klanke, C.; Heinkelein, M.; Rammling, M.; Dandekar, T.; von Kalle, C.; Rethwilm, A. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 2006, 87, 1339–1347. [Google Scholar]
- Deyle, D.R.; Khan, I.F.; Ren, G.; Russell, D.W. Lack of genotoxicity due to foamy virus vector integration in human iPSCs. Gene Ther. 2013. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar]
- Heneine, W.; Schweizer, M.; Sandstrom, P.; Folks, T. Human infection with foamy viruses. Curr. Top. Microbiol. Immunol. 2003, 277, 181–196. [Google Scholar]
- Hill, C.L.; Bieniasz, P.D.; McClure, M.O. Properties of human foamy virus relevant to its development as a vector for gene therapy. J. Gen. Virol. 1999, 80, 2003–2009. [Google Scholar]
- Rethwilm, A. Foamy virus vectors: An awaited alternative to gammaretro- and lentiviral vectors. Curr. Gene Ther. 2007, 7, 261–271. [Google Scholar]
- Liu, W.; Löchelt, M. 2008.
- Schmidt, M.; Niewiesk, S.; Heeney, J.; Aguzzi, A.; Rethwilm, A. Mouse model to study the replication of primate foamy viruses. J. Gen. Virol. 1997, 78, 1929–1933. [Google Scholar]
- Blochmann, R.; Coulibaly, C.; Bannert, N.; Cichutek, K.; Kurth, R.; Norley, S.; Fiebig, U. A novel small animal model to study the replication of simian foamy virus in vivo. In Proceedings of the 9th International Foamy Virus Conference 2012, Bethesda, MD, USA, 29 May 2012.
- Baunach, G.; Maurer, B.; Hahn, H.; Kranz, M.; Rethwilm, A. Functional analysis of human foamy virus accessory reading frames. J. Virol. 1993, 67, 5411–5418. [Google Scholar]
- Epstein, M.A. Simian retroviral infections in human beings. Lancet 2004, 364, 138–139; author reply 139–140. [Google Scholar] [CrossRef]
- Schmidt, M.; Herchenröder, O.; Heeney, J.; Rethwilm, A. Long terminal repeat U3 length polymorphism of human foamy virus. Virology 1997, 230, 167–178. [Google Scholar] [CrossRef]
- Herchenröder, O.; Renne, R.; Loncar, D.; Cobb, E.K.; Murthy, K.K.; Schneider, J.; Mergia, A.; Luciw, P.A. Isolation, cloning, and sequencing of simian foamy viruses from chimpanzees (SFVcpz): High homology to human foamy virus (HFV). Virology 1994, 201, 187–199. [Google Scholar] [CrossRef]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef]
- Münk, C.; Hechler, T.; Chareza, S.; Löchelt, M. Restriction of feline retroviruses: Lessons from cat APOBEC3 cytidine deaminases and TRIM5alpha proteins. Vet. Immunol. Immunopathol. 2010, 134, 14–24. [Google Scholar] [CrossRef]
- Alke, A.; Schwantes, A.; Zemba, M.; Flügel, R.M.; Löchelt, M. Characterization of the humoral immune response and virus replication in cats experimentally infected with feline foamy virus. Virology 2000, 275, 170–176. [Google Scholar] [CrossRef]
- Schwantes, A.; Ortlepp, I.; Löchelt, M. Construction and functional characterization of feline foamy virus-based retroviral vectors. Virology 2002, 301, 53–63. [Google Scholar] [CrossRef]
- Hatama, S.; Otake, K.; Omoto, S.; Murase, Y.; Ikemoto, A.; Mochizuki, M.; Takahashi, E.; Okuyama, H.; Fujii, Y. Isolation and sequencing of infectious clones of feline foamy virus and a human/feline foamy virus Env chimera. J. Gen. Virol. 2001, 82, 2999–3004. [Google Scholar]
- Wongsrikeao, P.; Saenz, D.; Rinkoski, T.; Otoi, T.; Poeschla, E. Antiviral restriction factor transgenesis in the domestic cat. Nat. Methods 2011, 8, 853–859. [Google Scholar] [CrossRef]
- Elder, J.H.; Phillips, T.R. Feline immunodeficiency virus as a model for development of molecular approaches to intervention strategies against lentivirus infections. Adv. Virus Res. 1995, 45, 225–247. [Google Scholar] [CrossRef]
- Roy, J.; Rudolph, W.; Juretzek, T.; Gartner, K.; Bock, M.; Herchenröder, O.; Lindemann, D.; Heinkelein, M.; Rethwilm, A. Feline foamy virus genome and replication strategy. J. Virol. 2003, 77, 11324–11331. [Google Scholar] [CrossRef]
- Zemba, M.; Alke, A.; Bodem, J.; Winkler, I.G.; Flower, R.L.; Pfrepper, K.; Delius, H.; Flügel, R.M.; Löchelt, M. Construction of infectious feline foamy virus genomes: Cat antisera do not cross-neutralize feline foamy virus chimera with serotype-specific Env sequences. Virology 2000, 266, 150–156. [Google Scholar] [CrossRef]
- Winkler, I.; Bodem, J.; Haas, L.; Zemba, M.; Delius, H.; Flower, R.; Flügel, R.M.; Löchelt, M. Characterization of the genome of feline foamy virus and its proteins shows distinct features different from those of primate spumaviruses. J. Virol. 1997, 71, 6727–6741. [Google Scholar]
- Weikel, J.; Löchelt, M.; Truyen, U. Demonstration of feline foamy virus in experimentally infected cats by immunohistochemistry. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2003, 50, 415–417. [Google Scholar] [CrossRef]
- Bastone, P.; Romen, F.; Liu, W.; Wirtz, R.; Koch, U.; Josephson, N.; Langbein, S.; Löchelt, M. Construction and characterization of efficient, stable and safe replication-deficient foamy virus vectors. Gene Ther. 2007, 14, 613–620. [Google Scholar] [CrossRef]
- Butera, S.T.; Brown, J.; Callahan, M.E.; Owen, S.M.; Matthews, A.L.; Weigner, D.D.; Chapman, L.E.; Sandstrom, P.A. Survey of veterinary conference attendees for evidence of zoonotic infection by feline retroviruses. J. Am. Vet. Med. Assoc. 2000, 217, 1475–1479. [Google Scholar] [CrossRef]
- von Laer, D.; Neumann-Haefelin, D.; Heeney, J.L.; Schweizer, M. Lymphocytes are the major reservoir for foamy viruses in peripheral blood. Virology 1996, 221, 240–244. [Google Scholar] [CrossRef]
- Helps, C.R.; Harbour, D.A. Comparison of the complete sequence of feline spumavirus with those of the primate spumaviruses reveals a shorter gag gene. J. Gen. Virol. 1997, 78, 2549–2564. [Google Scholar]
- Wirtz, R.; Löchelt, M. Construction and analysis of genomic, full-length infectious foamy virus DNA clones. Methods Mol. Biol. 2005, 304, 423–434. [Google Scholar]
- Bodem, J.; Kang, Y.; Flügel, R.M. Comparative functional characterization of the feline foamy virus transactivator reveals its species specificity. Virology 2004, 318, 32–36. [Google Scholar] [CrossRef]
- Löchelt, M.; Muranyi, W.; Flügel, R.M. Human foamy virus genome possesses an internal, Bel-1-dependent and functional promoter. Proc. Natl. Acad. Sci. USA 1993, 90, 7317–7321. [Google Scholar] [CrossRef]
- Löchelt, M.; Flügel, R.M.; Aboud, M. The human foamy virus internal promoter directs the expression of the functional Bel 1 transactivator and Bet protein early after infection. J. Virol. 1994, 68, 638–645. [Google Scholar]
- Löchelt, M.; Yu, S.F.; Linial, M.L.; Flügel, R.M. The human foamy virus internal promoter is required for efficient gene expression and infectivity. Virology 1995, 206, 601–610. [Google Scholar] [CrossRef]
- Lindemann, D.; Rethwilm, A. Characterization of a human foamy virus 170-kilodalton Env-Bet fusion protein generated by alternative splicing. J. Virol. 1998, 72, 4088–4094. [Google Scholar]
- Lecellier, C.H.; Vermeulen, W.; Bachelerie, F.; Giron, M.L.; Saib, A. Intra- and intercellular trafficking of the foamy virus auxiliary bet protein. J. Virol. 2002, 76, 3388–3394. [Google Scholar] [CrossRef]
- Giron, M.L.; de The, H.; Saib, A. An evolutionarily conserved splice generates a secreted env-Bet fusion protein during human foamy virus infection. J. Virol. 1998, 72, 4906–4910. [Google Scholar]
- Bodem, J.; Löchelt, M.; Delius, H.; Flügel, R.M. Detection of subgenomic cDNAs and mapping of feline foamy virus mRNAs reveals complex patterns of transcription. Virology 1998, 244, 417–426. [Google Scholar] [CrossRef]
- Alke, A.; Schwantes, A.; Kido, K.; Flötenmeyer, M.; Flügel, R.M.; Löchelt, M. The bet gene of feline foamy virus is required for virus replication. Virology 2001, 287, 310–320. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Chareza, S.; Slavkovic Lukic, D.; Liu, Y.; Räthe, A.M.; Münk, C.; Zabogli, E.; Pistello, M.; Löchelt, M. Molecular and functional interactions of cat APOBEC3 and feline foamy and immunodeficiency virus proteins: different ways to counteract host-encoded restriction. Virology 2012, 424, 138–146. [Google Scholar] [CrossRef]
- Perkovic, M.; Schmidt, S.; Marino, D.; Russell, R.A.; Stauch, B.; Hofmann, H.; Kopietz, F.; Kloke, B.P.; Zielonka, J.; Strover, H.; et al. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 2009, 284, 5819–5826. [Google Scholar]
- Münk, C.; Beck, T.; Zielonka, J.; Hotz-Wagenblatt, A.; Chareza, S.; Battenberg, M.; Thielebein, J.; Cichutek, K.; Bravo, I.G.; O'Brien, S.J.; et al. Functions, structure, and read-through alternative splicing of feline APOBEC3 genes. Genome Biol. 2008, 9, R48. [Google Scholar] [CrossRef]
- Münk, C.; Zielonka, J.; Constabel, H.; Kloke, B.P.; Rengstl, B.; Battenberg, M.; Bonci, F.; Pistello, M.; Löchelt, M.; Cichutek, K. Multiple restrictions of human immunodeficiency virus type 1 in feline cells. J. Virol. 2007, 81, 7048–7060. [Google Scholar] [CrossRef]
- Dawson, S.; Bennett, D.; Carter, S.D.; Bennett, M.; Meanger, J.; Turner, P.C.; Carter, M.J.; Milton, I.; Gaskell, R.M. Acute arthritis of cats associated with feline calicivirus infection. Res. Vet. Sci. 1994, 56, 133–143. [Google Scholar] [CrossRef]
- Hoover, E.A.; Kahn, D.E. Experimentally induced feline calicivirus infection: clinical signs and lesions. J. Am. Vet. Med. Assoc. 1975, 166, 463–468. [Google Scholar]
- Knowles, J.O.; McArdle, F.; Dawson, S.; Carter, S.D.; Gaskell, C.J.; Gaskell, R.M. Studies on the role of feline calicivirus in chronic stomatitis in cats. Vet. Microbiol. 1991, 27, 205–219. [Google Scholar] [CrossRef]
- Geissler, K.; Schneider, K.; Platzer, G.; Truyen, B.; Kaaden, O.R.; Truyen, U. Genetic and antigenic heterogeneity among feline calicivirus isolates from distinct disease manifestations. Virus Res. 1997, 48, 193–206. [Google Scholar] [CrossRef]
- Geissler, K.; Schneider, K.; Truyen, U. Mapping neutralizing and non-neutralizing epitopes on the capsid protein of feline calicivirus. J. Vet. Med. B Infect. Dis. Vet. Public Health 2002, 49, 55–60. [Google Scholar] [CrossRef]
- Liu, W.; Backes, P.; Löchelt, M. Importance of the major splice donor and redefinition of cis-acting sequences of gutless feline foamy virus vectors. Virology 2009, 394, 208–217. [Google Scholar] [CrossRef]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar]
- Schmidt, M.; Rethwilm, A. Replicating foamy virus-based vectors directing high level expression of foreign genes. Virology 1995, 210, 167–178. [Google Scholar] [CrossRef]
- Yap, M.W.; Lindemann, D.; Stanke, N.; Reh, J.; Westphal, D.; Hanenberg, H.; Ohkura, S.; Stoye, J.P. Restriction of foamy viruses by primate Trim5alpha. J. Virol. 2008, 82, 5429–5439. [Google Scholar]
- Bastone, P.; Löchelt, M. Kinetics and characteristics of replication-competent revertants derived from self-inactivating foamy virus vectors. Gene Ther. 2004, 11, 465–473. [Google Scholar] [CrossRef]
- Cooper, D.N.; Gerber-Huber, S. DNA methylation and CpG suppression. Cell Differ. 1985, 17, 199–205. [Google Scholar]
- Bastone, P.; Bravo, I.G.; Löchelt, M. Feline foamy virus-mediated marker gene transfer: Identification of essential genetic elements and influence of truncated and chimeric proteins. Virology 2006, 348, 190–199. [Google Scholar] [CrossRef]
- Heinkelein, M.; Dressler, M.; Jarmy, G.; Rammling, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Rethwilm, A. Improved primate foamy virus vectors and packaging constructs. J. Virol. 2002, 76, 3774–3783. [Google Scholar] [CrossRef]
- Erlwein, O.; Bieniasz, P.D.; McClure, M.O. Sequences in pol are required for transfer of human foamy virus-based vectors. J. Virol. 1998, 72, 5510–5516. [Google Scholar]
- Heinkelein, M.; Schmidt, M.; Fischer, N.; Moebes, A.; Lindemann, D.; Enssle, J.; Rethwilm, A. Characterization of a cis-acting sequence in the Pol region required to transfer human foamy virus vectors. J. Virol. 1998, 72, 6307–6314. [Google Scholar]
- Wu, M.; Chari, S.; Yanchis, T.; Mergia, A. cis-Acting sequences required for simian foamy virus type 1 vectors. J. Virol. 1998, 72, 3451–3454. [Google Scholar]
- Peters, K.; Wiktorowicz, T.; Heinkelein, M.; Rethwilm, A. RNA and protein requirements for incorporation of the Pol protein into foamy virus particles. J. Virol. 2005, 79, 7005–7013. [Google Scholar] [CrossRef]
- Wiktorowicz, T.; Peters, K.; Armbruster, N.; Steinert, A.F.; Rethwilm, A. Generation of an improved foamy virus vector by dissection of cis-acting sequences. J. Gen. Virol. 2009, 90, 481–487. [Google Scholar] [CrossRef]
- Heinkelein, M.; Thurow, J.; Dressler, M.; Imrich, H.; Neumann-Haefelin, D.; McClure, M.O.; Rethwilm, A. Complex effects of deletions in the 5' untranslated region of primate foamy virus on viral gene expression and RNA packaging. J. Virol. 2000, 74, 3141–3148. [Google Scholar] [CrossRef]
- Pietschmann, T.; Heinkelein, M.; Heldmann, M.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. Foamy virus capsids require the cognate envelope protein for particle export. J. Virol. 1999, 73, 2613–2621. [Google Scholar]
- Ho, Y.P.; Schnabel, V.; Swiersy, A.; Stirnnagel, K.; Lindemann, D. A small-molecule-controlled system for efficient pseudotyping of prototype foamy virus vectors. Mol. Ther. 2012, 20, 1167–1176. [Google Scholar] [CrossRef]
- Plochmann, K.; Horn, A.; Gschmack, E.; Armbruster, N.; Krieg, J.; Wiktorowicz, T.; Weber, C.; Stirnnagel, K.; Lindemann, D.; Rethwilm, A.; et al. Heparan sulfate is an attachment factor for foamy virus entry. J. Virol. 2012, 86, 10028–10035. [Google Scholar]
- Burton, D.R.; Weiss, R.A. AIDS/HIV. A boost for HIV vaccine design. Science 2010, 329, 770–773. [Google Scholar] [CrossRef]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.Y.; Dai, K.; Finzi, A.; Kwon, Y.D.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 2010, 329, 811–817. [Google Scholar] [CrossRef]
- Pejchal, R.; Walker, L.M.; Stanfield, R.L.; Phogat, S.K.; Koff, W.C.; Poignard, P.; Burton, D.R.; Wilson, I.A. Structure and function of broadly reactive antibody PG16 reveal an H3 subdomain that mediates potent neutralization of HIV-1. Proc. Natl. Acad. Sci. USA 2010, 107, 11483–11488. [Google Scholar]
- Talbott, R.L.; Sparger, E.E.; Lovelace, K.M.; Fitch, W.M.; Pedersen, N.C.; Luciw, P.A.; Elder, J.H. Nucleotide sequence and genomic organization of feline immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1989, 86, 5743–5747. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W.; et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef]
- Bleiholder, A.; Löchelt, M. 2012.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, W.; Lei, J.; Liu, Y.; Lukic, D.S.; Räthe, A.-M.; Bao, Q.; Kehl, T.; Bleiholder, A.; Hechler, T.; Löchelt, M. Feline Foamy Virus-Based Vectors: Advantages of an Authentic Animal Model. Viruses 2013, 5, 1702-1718. https://doi.org/10.3390/v5071702
Liu W, Lei J, Liu Y, Lukic DS, Räthe A-M, Bao Q, Kehl T, Bleiholder A, Hechler T, Löchelt M. Feline Foamy Virus-Based Vectors: Advantages of an Authentic Animal Model. Viruses. 2013; 5(7):1702-1718. https://doi.org/10.3390/v5071702
Chicago/Turabian StyleLiu, Weibin, Janet Lei, Yang Liu, Dragana Slavkovic Lukic, Ann-Mareen Räthe, Qiuying Bao, Timo Kehl, Anne Bleiholder, Torsten Hechler, and Martin Löchelt. 2013. "Feline Foamy Virus-Based Vectors: Advantages of an Authentic Animal Model" Viruses 5, no. 7: 1702-1718. https://doi.org/10.3390/v5071702
APA StyleLiu, W., Lei, J., Liu, Y., Lukic, D. S., Räthe, A.-M., Bao, Q., Kehl, T., Bleiholder, A., Hechler, T., & Löchelt, M. (2013). Feline Foamy Virus-Based Vectors: Advantages of an Authentic Animal Model. Viruses, 5(7), 1702-1718. https://doi.org/10.3390/v5071702