Role of Polycomb Proteins in Regulating HSV-1 Latency

Abstract
:1. Introduction
1.1. Evidence for the Role of Histone Post-Translational Modifications (PTMs) in Regulating Lytic and Latent Phases of Infection
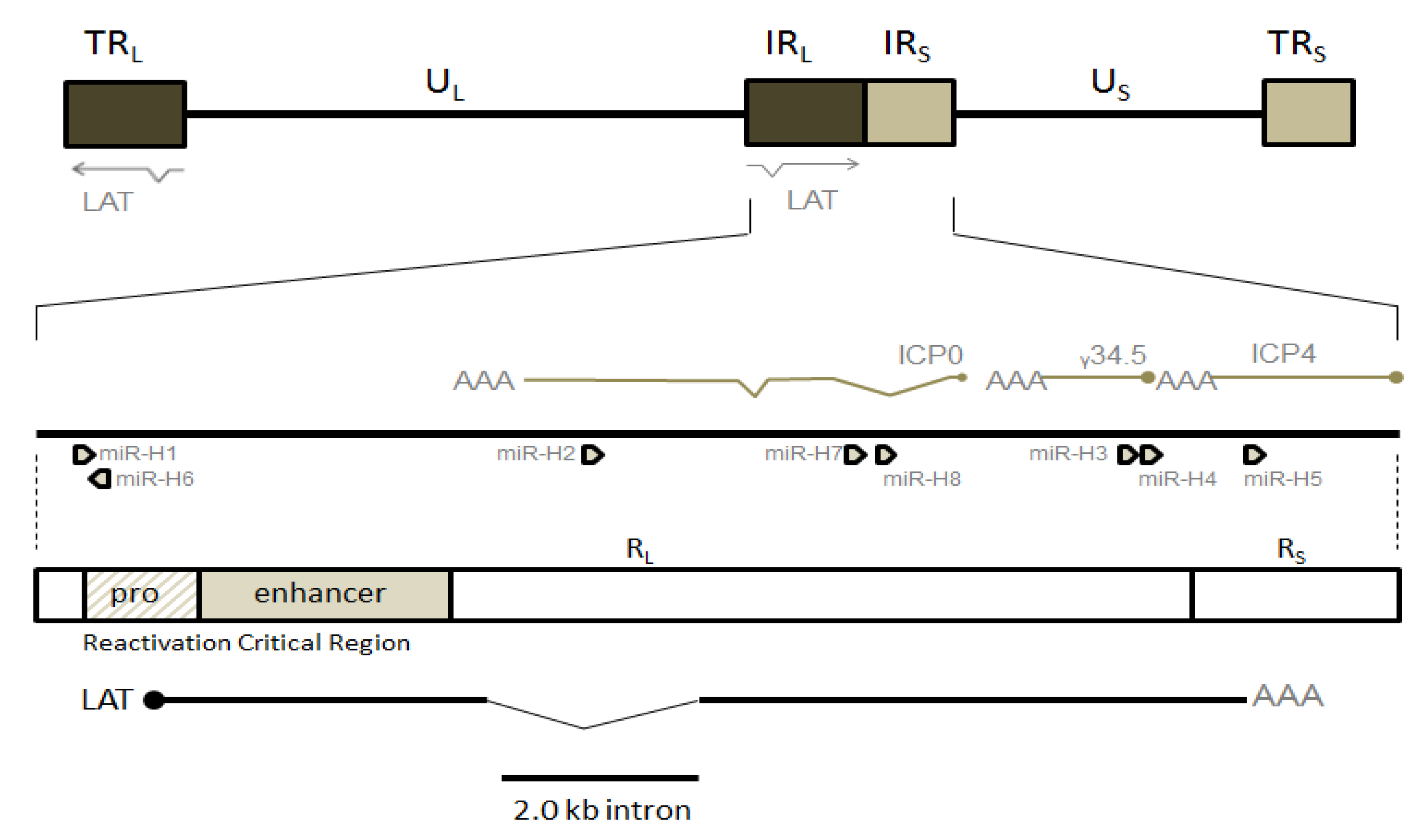
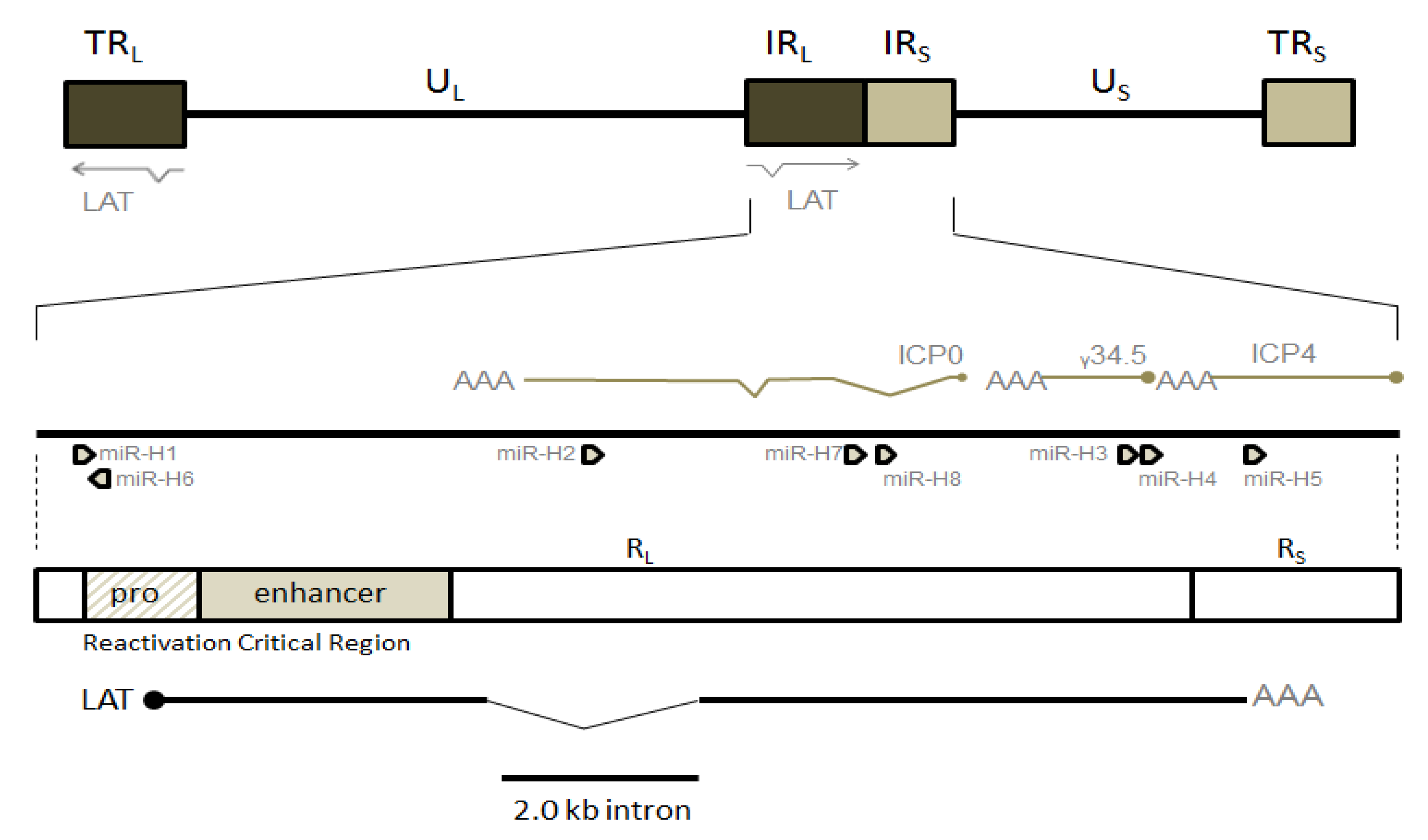
1.2. Animal Models Used to Study HSV-1 Latency
1.3. In Vitro Models of HSV-1 Latency
1.4. Differences in Biological Properties of Different Strains Used to Study HSV-1 Latency
2. Neuronal Basis of HSV Latency
3. Chromatinization of HSV-1 Genomes during Latency
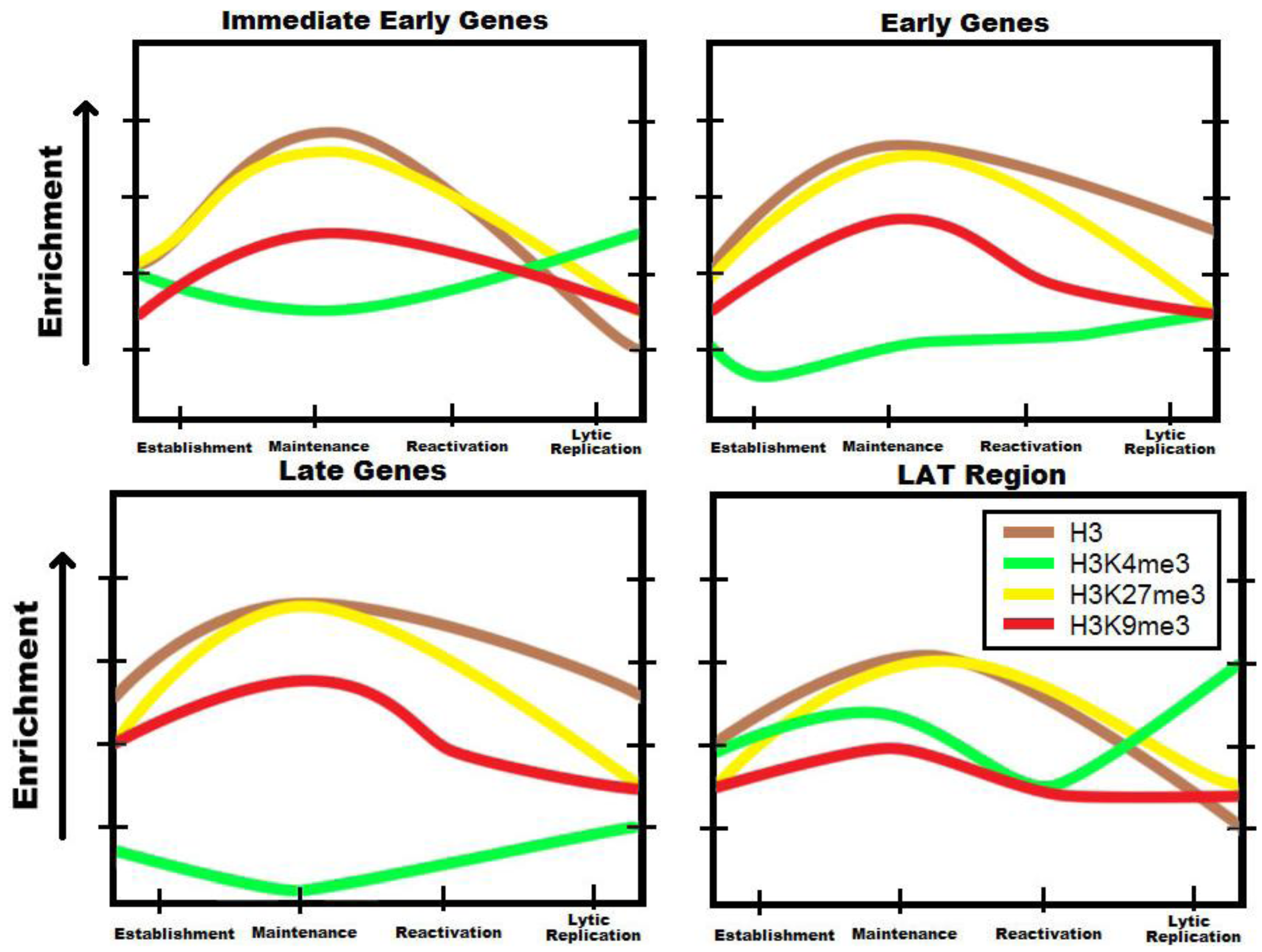
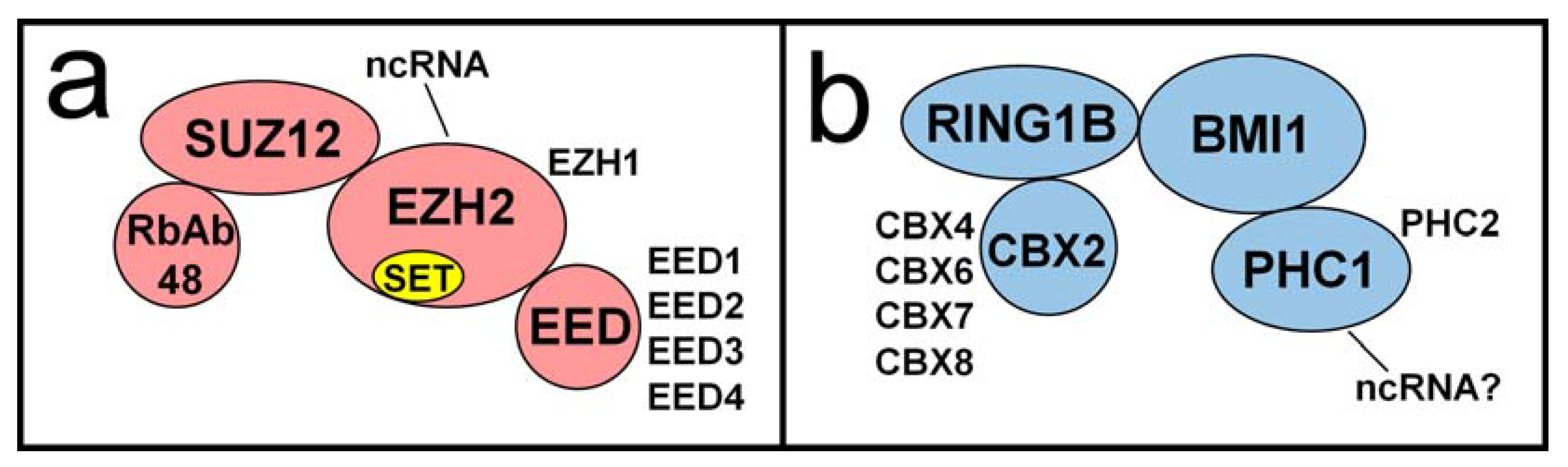
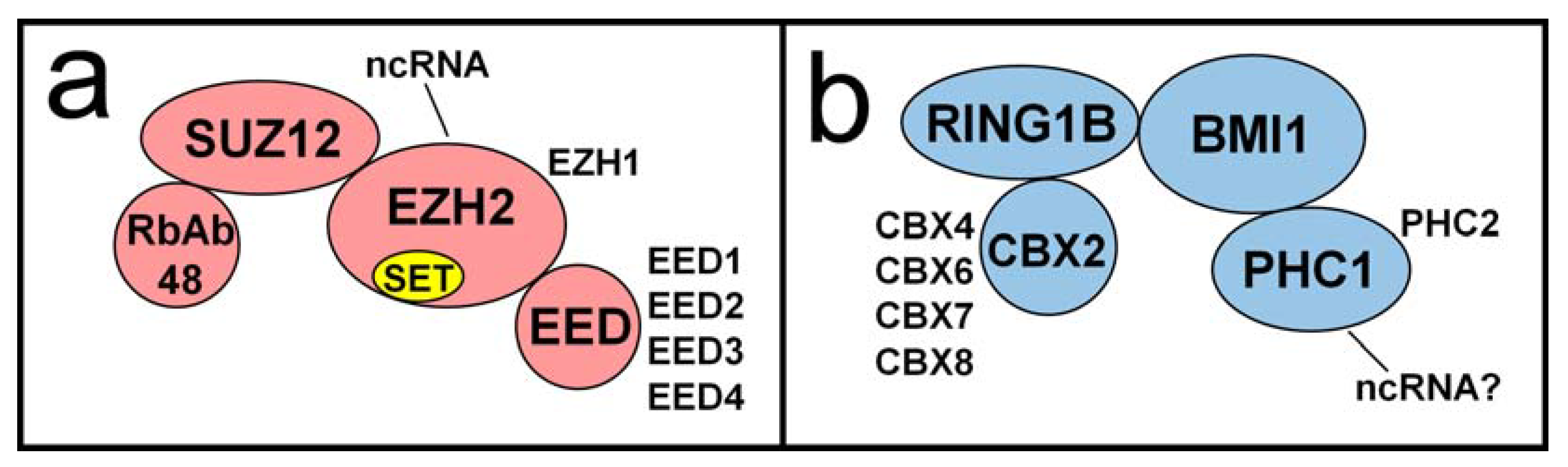
4. Polycomb Proteins Role in Regulating Lytic Gene Activity during Latency
5. The Predicted Role of Histone Demethylases in HSV-1 Reactivation

{kind=link}
{kind=link}
{kind=link}
| Demethylase | Family | Specificity | Associated Complex | Biological Role | Inhibitor |
|---|---|---|---|---|---|
| LSD1 (KDM1A) | FAD-amine oxidase | H3K4me2/me1 H3K9me2/me1 | HDAC1/ CoREST/ REST | Possible coordinated role with HDACs in transcription repression | Paraglyine a, TCP, OG-L002 |
| JHD3A/JMJD2A (KDM4A) JHD3C/JMJD2C/ GASC1 (KDM4C) | Jumonji b | H3K9me3/me2 H3K36me3/me2 H1.4K26me c | NCoR complex d | KDM4C is a possible oncogene e | PCA, NOG, DMOG, ML324 |
| JARID1A/RBP2 (KDM5A) JARID1C/SMCX (KDM5C) | Jumonji | H3K4me3/me2 | Sin3/HDAC complex (KDM5A) NCoR/REST (KDM5C) | Notch signaling (JARID1A) NCoR-SMCX-REST complex functions in glial development | N/A f |
| UTX (KDM6A) | Jumonji | H3K27me3 | MLL3/4, RbBP5, WDR5, and ASH2 | Pluripotent stem cell differentiation (Hox gene regulation) | GSK-J4 |
| JMJD3(KDM6B) | Jumonji | H3K27me3 | RbBP5 | Induced upon activation of macrophages by inflammatory stimuli Role in neuronal commitment | GSK-J4 |
5.1. FAD-Amine Oxidase: Lysine Specific Demethylase 1 (LSD1/KDM1)
5.2. Jumonji-Domain Histone Demethylases: JMJD3 (KDM6B) and UTX (KDM6A)
6. Summary and Discussion
Acknowledgments
Conflict of Interest
References and Notes
- Gressens, P.; Martin, J.R. In situ polymerase chain reaction: Localization of HSV-2 DNA sequences in infections of the nervous system. J. Virol. Methods 1994, 46, 61–83. [Google Scholar] [CrossRef]
- Mehta, A.; Maggioncalda, J.; Bagasra, O.; Thikkavarapu, S.; Saikumari, P.; Valyi-Nagy, T.; Fraser, N.W.; Block, T.M. In situ DNA PCR and RNA hybridization of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virology 1995, 206, 633–640. [Google Scholar] [CrossRef]
- Stevens, J.G.; Wagner, E.K.; Devi, R.G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar]
- Farrell, M.J.; Dobson, A.T.; Feldman, L.T. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA 1991, 88, 790–794. [Google Scholar] [CrossRef]
- Wechsler, S.L.; Nesburn, A.B.; Watson, R.; Slanina, S.M.; Ghiasi, H. Fine mapping of the latency-related gene of herpes simplex virus type 1: Alternative splicing produces distinct latency-related RNAs containing open reading frames. J. Virol. 1988, 62, 4051–4058. [Google Scholar]
- Sawtell, N.M.; Thompson, R.L. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J. Virol. 1992, 66, 2150–2156. [Google Scholar]
- Shimeld, C.; Hill, T.J.; Blyth, W.A.; Easty, D.L. Reactivation of latent infection and induction of recurrent herpetic eye disease in mice. J. Gen. Virol. 1990, 71, 397–404. [Google Scholar] [CrossRef]
- Neumann, D.M.; Bhattacharjee, P.S.; Giordani, N.V.; Bloom, D.C.; Hill, J.M. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J. Virol. 2007, 81, 13248–13253. [Google Scholar] [CrossRef]
- Sawtell, N.M.; Thompson, R.L. Comparison of herpes simplex virus reactivation in ganglia in vivo and in explants demonstrates quantitative and qualitative differences. J. Virol. 2004, 78, 7784–7794. [Google Scholar] [CrossRef]
- Wilson, A.C.; Mohr, I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 2012, 20, 604–611. [Google Scholar] [CrossRef]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, V.; Neumann, D.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J. Biomed. Biotechnol. 2012, 2012, 612316. [Google Scholar]
- Herrera, F.J.; Triezenberg, S.J. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 2004, 78, 9689–9696. [Google Scholar] [CrossRef]
- Huang, J.; Kent, J.R.; Placek, B.; Whelan, K.A.; Hollow, C.M.; Zeng, P.Y.; Fraser, N.W.; Berger, S.L. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J. Virol. 2006, 80, 5740–5746. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef]
- Kutluay, S.B.; Triezenberg, S.J. Regulation of histone deposition on the herpes simplex virus type 1 genome during lytic infection. J. Virol. 2009, 83, 5835–5845. [Google Scholar] [CrossRef]
- Hancock, M.H.; Cliffe, A.R.; Knipe, D.M.; Smiley, J.R. Herpes simplex virus VP16, but not ICP0, is required to reduce histone occupancy and enhance histone acetylation on viral genomes in U2OS osteosarcoma cells. J. Virol. 2010, 84, 1366–1375. [Google Scholar] [CrossRef]
- Kent, J.R.; Zeng, P.Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar]
- Oh, J.; Fraser, N.W. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 2008, 82, 3530–3537. [Google Scholar] [CrossRef]
- Coleman, H.M.; Connor, V.; Cheng, Z.S.; Grey, F.; Preston, C.M.; Efstathiou, S. Histone modifications associated with herpes simplex virus type 1 genomes during quiescence and following ICP0-mediated de-repression. J. Gen. Virol. 2008, 89, 68–77. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; DeLuca, N.A. Reversal of heterochromatic silencing of quiescent herpes simplex virus type 1 by ICP0. J. Virol. 2011, 85, 3424–3435. [Google Scholar] [CrossRef]
- Preston, C.M.; Efstathiou, S. Molecular Basis of HSV Latency and Reactivation. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Bertke, A.S.; Swanson, S.M.; Chen, J.; Imai, Y.; Kinchington, P.R.; Margolis, T.P. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 2011, 85, 6669–6677. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kim, J.Y.; Camarena, V.; Roehm, P.C.; Chao, M.V.; Wilson, A.C.; Mohr, I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 2012, 62. [Google Scholar] [CrossRef]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar]
- Giordani, N.V.; Neumann, D.M.; Kwiatkowski, D.L.; Bhattacharjee, P.S.; McAnany, P.K.; Hill, J.M.; Bloom, D.C. During HSV-1 infection of rabbits, the ability to express the LAT increases latent-phase transcription of lytic genes. J. Virol. 2008, 82, 6056–6060. [Google Scholar] [CrossRef]
- Perng, G.C.; Esmaili, D.; Slanina, S.M.; Yukht, A.; Ghiasi, H.; Osorio, N.; Mott, K.R.; Maguen, B.; Jin, L.; Nesburn, A.B.; et al. Three herpes simplex virus type 1 latency-associated transcript mutants with distinct and asymmetric effects on virulence in mice compared with rabbits. J. Virol. 2001, 75, 9018–9028. [Google Scholar] [CrossRef]
- Sawtell, N.M.; Thompson, R.L. Herpes simplex virus type 1 latency associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J. Virol. 1992, 66, 2157–2169. [Google Scholar]
- Kolokotronis, A.; Doumas, S. Herpes simplex virus infection, with particular reference to the progression and complications of primary herpetic gingivostomatitis. Clin. Microbiol. Infect. 2006, 12, 202–211. [Google Scholar] [CrossRef]
- LaVail, J.H.; Zhan, J.; Margolis, T.P. HSV (type 1) infection of the trigeminal complex. Brain Res. 1990, 514, 181–188. [Google Scholar] [CrossRef]
- Raible, D.W.; Ungos, J.M. Specification of sensory neuron cell fate from the neural crest. Adv. Exp. Med. Biol. 2006, 589, 170–180. [Google Scholar] [CrossRef]
- Margolis, T.P.; Dawson, C.R.; LaVail, J.H. Herpes simplex viral infection of the mouse trigeminal ganglion. Immunohistochemical analysis of cell populations. Invest. Ophthalmol. Vis. Sci. 1992, 33, 259–267. [Google Scholar]
- Yang, L.; Voytek, C.C.; Margolis, T.P. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J. Virol. 2000, 74, 209–217. [Google Scholar] [CrossRef]
- Margolis, T.P.; Sedarati, F.; Dobson, A.T.; Feldman, L.T.; Stevens, J.G. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology 1992, 189, 150–160. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes-simplex virus Type-1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar]
- Lacasse, J.J.; Schang, L.M. Herpes simplex virus 1 DNA is in unstable nucleosomes throughout the lytic infection cycle, and the instability of the nucleosomes is independent of DNA replication. J. Virol. 2012, 86, 11287–11300. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Quina, A.S.; Buschbeck, M.; Di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef]
- Bartova, E.; Krejci, J.; Harnicarova, A.; Galiova, G.; Kozubek, S. Histone modifications and nuclear architecture: A review. J. Histochem. Cytochem. 2008, 56, 711–721. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Coen, D.M.; Knipe, D.M. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. MBio 2013, 4. [Google Scholar] [CrossRef]
- Kubat, N.J.; Tran, R.K.; McAnany, P.; Bloom, D.C. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 2004, 78, 1139–1149. [Google Scholar] [CrossRef]
- Kubat, N.J.; Amelio, A.L.; Giordani, N.V.; Bloom, D.C. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J. Virol. 2004, 78, 12508–12518. [Google Scholar] [CrossRef]
- Amelio, A.L.; Giordani, N.V.; Kubat, N.J.; O’Neil, J.E.; Bloom, D.C. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J. Virol. 2006, 80, 2063–2068. [Google Scholar] [CrossRef]
- Creech, C.C.; Neumann, D.M. Changes to euchromatin on LAT and ICP4 following reactivation are more prevalent in an efficiently reactivating strain of HSV-1. PLoS One 2010, 5, e15416. [Google Scholar] [CrossRef]
- St Leger, A.J.; Hendricks, R.L. CD8+ T cells patrol HSV-1-infected trigeminal ganglia and prevent viral reactivation. J. Neurovirol. 2011, 17, 528–534. [Google Scholar] [CrossRef]
- Dressler, G.R.; Rock, D.L.; Fraser, N.W. Latent herpes simplex virus type 1 DNA is not extensively methylated in vivo. J. Gen. Virol. 1987, 68, 1761–1765. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef]
- Oktaba, K.; Gutierrez, L.; Gagneur, J.; Girardot, C.; Sengupta, A.K.; Furlong, E.E.; Muller, J. Dynamic regulation by polycomb group protein complexes controls pattern formation and the cell cycle in Drosophila. Dev. Cell 2008, 15, 877–889. [Google Scholar] [CrossRef]
- Ringrose, L.; Paro, R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 2007, 134, 223–232. [Google Scholar] [CrossRef]
- Cao, D.; Wang, Z.; Zhang, C.L.; Oh, J.; Xing, W.; Li, S.; Richardson, J.A.; Wang, D.Z.; Olson, E.N. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol. Cell Biol. 2005, 25, 364–376. [Google Scholar] [CrossRef]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef]
- Cao, R.; Tsukada, Y.; Zhang, Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and hox gene silencing. Mol. Cell 2005, 20, 845–854. [Google Scholar] [CrossRef]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin compaction by a polycomb group protein complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef]
- Chopra, V.S.; Hendrix, D.A.; Core, L.J.; Tsui, C.; Lis, J.T.; Levine, M. The polycomb group mutant esc leads to augmented levels of paused Pol II in the Drosophila embryo. Mol. Cell 2011, 42, 837–844. [Google Scholar] [CrossRef]
- Fischle, W.; Wang, Y.; Jacobs, S.A.; Kim, Y.; Allis, C.D.; Khorasanizadeh, S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003, 17, 1870–1881. [Google Scholar] [CrossRef]
- Margolis, T.P.; Elfman, F.L.; Leib, D.; Pakpour, N.; Apakupakul, K.; Imai, Y.; Voytek, C. Spontaneous reactivation of herpes simplex virus type 1 in latently infected murine sensory ganglia. J. Virol. 2007, 81, 11069–11074. [Google Scholar]
- Feldman, L.; Ellison, A.R.; Voytek, C.C.; Yang, L.; Krause, P.; Margolis, T.P. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 978–983. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef]
- Sing, A.; Pannell, D.; Karaiskakis, A.; Sturgeon, K.; Djabali, M.; Ellis, J.; Lipshitz, H.D.; Cordes, S.P. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell 2009, 138, 885–897. [Google Scholar] [CrossRef]
- Woo, C.J.; Kharchenko, P.V.; Daheron, L.; Park, P.J.; Kingston, R.E. A region of the human HOXD cluster that confers polycomb-group responsiveness. Cell 2010, 140, 99–110. [Google Scholar] [CrossRef]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef]
- Bloom, D.C.; Devi-Rao, G.B.; Hill, J.M.; Stevens, J.G.; Wagner, E.K. Molecular analysis of herpes simplex virus type 1 during epinephrine induced reactivation of latently infected rabbits in vivo. J. Virol. 1994, 68, 1283–1292. [Google Scholar]
- Maggioncalda, J.; Mehta, A.; Su, Y.H.; Fraser, N.W.; Block, T.M. Correlation between herpes simplex virus type 1 rate of reactivation from latent infection and the number of infected neurons in trigeminal ganglia. Virology 1996, 225, 72–81. [Google Scholar] [CrossRef]
- Hermanson, O.; Jepsen, K.; Rosenfeld, M.G. N-CoR controls differentiation of neural stem cells into astrocytes. Nature 2002, 419, 934–939. [Google Scholar] [CrossRef]
- Yang, Z.Q.; Imoto, I.; Fukuda, Y.; Pimkhaokham, A.; Shimada, Y.; Imamura, M.; Sugano, S.; Nakamura, Y.; Inazawa, J. Identification of a novel gene, GASC1, within an amplicon at 9p23–24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000, 60, 4735–4739. [Google Scholar]
- Gu, H.; Roizman, B. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 2009, 83, 4376–4385. [Google Scholar] [CrossRef]
- Shi, Y.J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 2005, 19, 857–864. [Google Scholar]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef]
- Liang, Y.; Quenelle, D.; Vogel, J.L.; Mascaro, C.; Ortega, A.; Kristie, T.M. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio 2013, 4. [Google Scholar] [CrossRef]
- Burgold, T.; Spreafico, F.; De Santa, F.; Totaro, M.G.; Prosperini, E.; Natoli, G.; Testa, G. The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS One 2008, 3, e3034. [Google Scholar] [CrossRef]
- De Santa, F.; Narang, V.; Yap, Z.H.; Tusi, B.K.; Burgold, T.; Austenaa, L.; Bucci, G.; Caganova, M.; Notarbartolo, S.; Casola, S.; et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 2009, 28, 3341–3352. [Google Scholar] [CrossRef]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Watson, Z.; Dhummakupt, A.; Messer, H.; Phelan, D.; Bloom, D. Role of Polycomb Proteins in Regulating HSV-1 Latency. Viruses 2013, 5, 1740-1757. https://doi.org/10.3390/v5071740
Watson Z, Dhummakupt A, Messer H, Phelan D, Bloom D. Role of Polycomb Proteins in Regulating HSV-1 Latency. Viruses. 2013; 5(7):1740-1757. https://doi.org/10.3390/v5071740
Chicago/Turabian StyleWatson, Zachary, Adit Dhummakupt, Harald Messer, Dane Phelan, and David Bloom. 2013. "Role of Polycomb Proteins in Regulating HSV-1 Latency" Viruses 5, no. 7: 1740-1757. https://doi.org/10.3390/v5071740