Molecular Phylogeny of Hantaviruses Harbored by Insectivorous Bats in Côte d’Ivoire and Vietnam

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
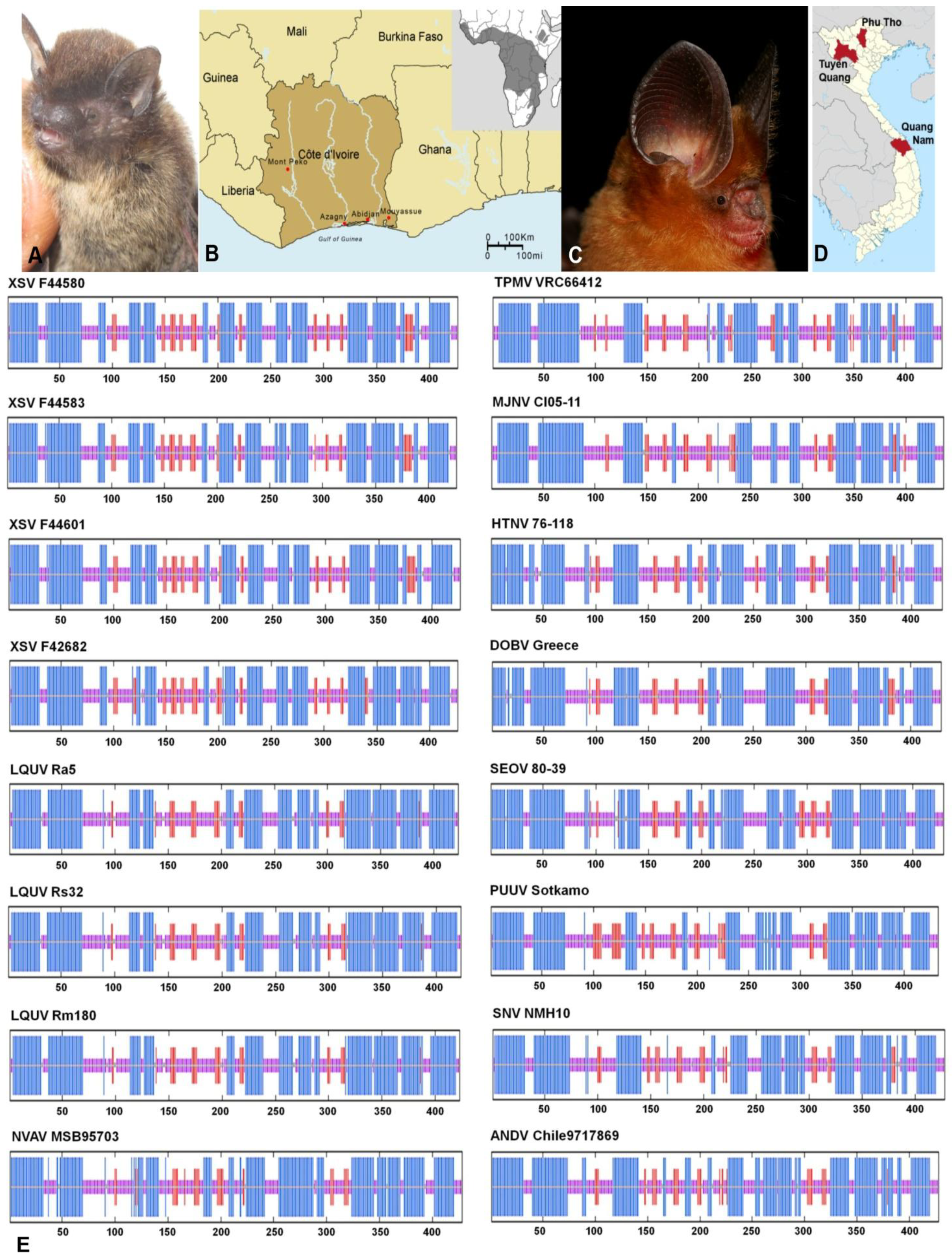
2.1. Hantavirus Detection and Sequence Analysis

{kind=link}
{kind=link}
{kind=link}
| Bat Family | Frozen | RNAlater® | Ethanol-fixed | Total | |||||
|---|---|---|---|---|---|---|---|---|---|
| Lung | Liver | Kidney | Lung | Intercostal Muscle | Intestine | Rectal Swab or Feces | Liver | ||
| Hipposideridae | 50 | 7 | 57 | ||||||
| Molossidae | 1 | 35 | 6 | 42 | |||||
| Nycteridae | 1 | 1 | 2 | ||||||
| Pteropodidae | 42 | 18 | 60 | ||||||
| Phyllostomidae | 2 | 2 | |||||||
| Rhinolophidae | 150 | 12 | 162 | ||||||
| Vespertilionidae | 11 | 17 | 1 | 49 | 45 | 73 | 12 | 208 | |
| Total | 164 | 17 | 51 | 146 | 19 | 45 | 79 | 12 | 533 |
| Virus | Strain | Bat Species | Country | Province | S | M | L |
|---|---|---|---|---|---|---|---|
| XSV | VN1982 | Hipposideros pomona | Vietnam | Phu Tho | 499 bp | 4582 bp | |
| KC688335 | JX912953 | ||||||
| F42640 | Tuyên Quang | 516 bp | 567 bp | ||||
| KF704708 | KF704713 | ||||||
| F42682 | 1752 bp | 663 bp | 1160 bp | ||||
| KF704709 | KJ000538 | KF704714 | |||||
| F44580 | Quang Nam | 1728 bp | 804 bp | ||||
| KF704710 | KF704715 | ||||||
| F44583 | 1728 bp | 1160 bp | |||||
| KF704711 | KF704716 | ||||||
| F44601 | 1728 bp | 663 bp | 1160 bp | ||||
| KF704712 | KJ000539 | KF704717 | |||||
| MOYV | KB576 | Neoromicia nanus | Côte d'Ivoire | Mouyassué | 1691 bp | ||
| JQ287716 | |||||||
| KB577 | 372 bp KJ000540 |


2.2. Nucleocapsid Secondary Structure
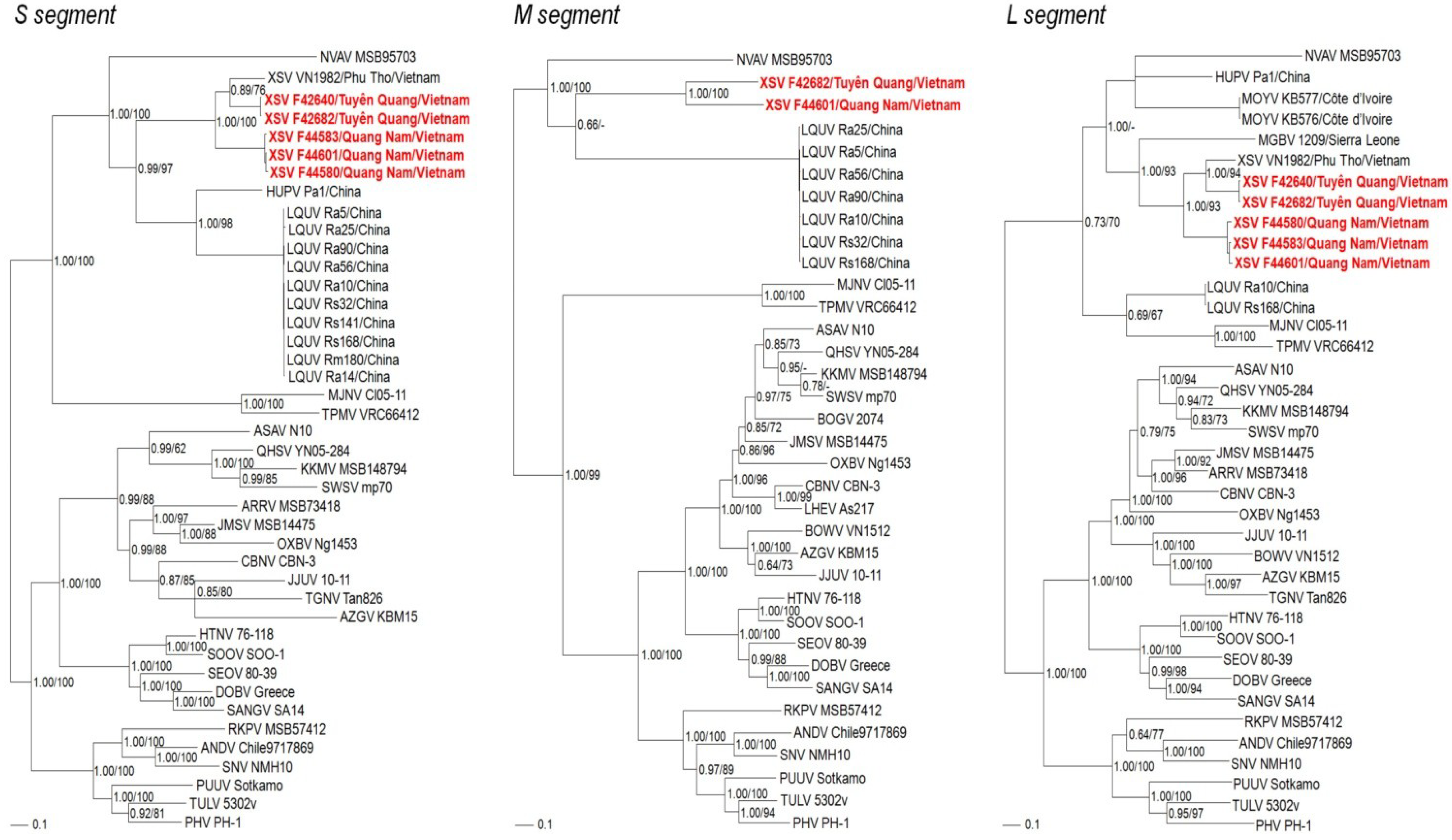
2.3. Phylogenetic Analysis

2.4. Bats as Hosts of Hantaviruses
3. Experimental Section
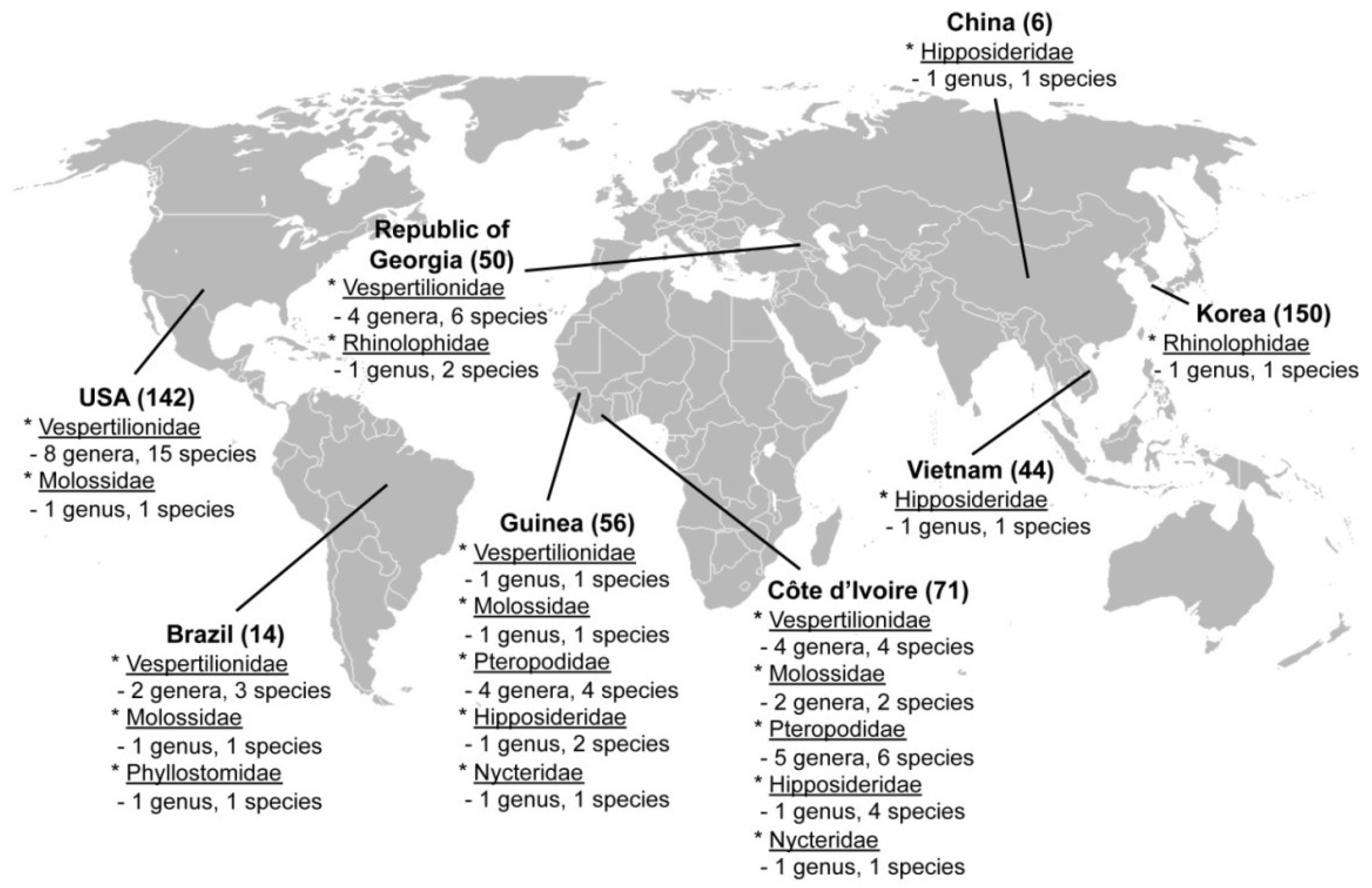
3.1. Samples
3.2. Genome Detection and Sequencing
| Primer | Sequence (5’-3’) | Segment | Polarity |
|---|---|---|---|
| Han-5’end-EcoRI | CTC GAA TTC TAG TAG TAG AC | S | + |
| Shrew-S777R | AAN CCD ATN ACN CCC AT | S | - |
| Shrew-S764R | CCA TNA CWG GRC TNA TCA | S | - |
| XSV-S627F | AGA AGA ATT GAC ACC TGG GCG AT | S | + |
| XSV-S1040F | CAT TCT TTT CAC TGT TGC AGG A | S | + |
| XSV-S1235R | GTT CTT CTG AGA TAT GAC TGA TA | S | - |
| Bat-3’endR | TAG TAG TAK RCT CCC T | S | - |
| G2F1 | TGG GCT GCA AGT GC | M | + |
| Han-M2957R | GAR CCC CAN GCN CCN TCW AT | M | - |
| Han-M2631R | CAT NAY RTC NCC RGG RTC NCC | M | - |
| Han-L1880F | CAR AAR ATG AAR NTN TGT GC | L | + |
| Bat-L1929F | ATG AAR NTN TGT GCA YTG TTT GA | L | + |
| Han-L2520F | ATN WGH YTD AAR GGN ATG TCN GG | L | + |
| Bat-L2810F | GAR GAY TAY TAT GAT G | L | + |
| Han-L3000R | GCN GAR TTR TCN CCN GGN GAC CA | L | - |
| Han-L2970R | CCN GGN GAC CAY TTN GTD GCA TC | L | - |
| MOYV-L2683R | GCT GGA TAA CAG TCG GGT TTA ATC | L | - |
| MOYV-L2612R | TAA GTG CCC ATC TTC TTG TA | L | - |
| Bat-L3442R | ACC ART CWG AMC CAT CAT C | L | - |
| Bat-L3613R | GTA GAG AGA AAC TCT GCA TTT GT | L | - |
3.3. Phylogenetic Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Plyusnin, A.; Vapalahti, O.; Vaheri, A. Hantaviruses: Genome structure, expression and evolution. J. Gen. Virol. 1996, 77, 2677–2687. [Google Scholar] [CrossRef]
- Plyusnin, A.; Beatty, B.J.; Elliott, R.M.; Goldbach, R.; Kormelink, R.; Lundkvist, A.; Schmaljohn, C.S.; Tesh, R.B. Bunyaviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses; King, A.M.Q., Lefkowitz, E.J., Adams, M.J., Carstens, E.B., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2012; pp. 725–741. [Google Scholar]
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Barriere, P.; Koivogui, L.; terMeulen, J.; Krüger, D.H. Novel hantavirus sequences in shrew, Guinea. Emerg. Infect. Dis. 2007, 13, 520–522. [Google Scholar] [CrossRef]
- Arai, S.; Song, J.W.; Sumibcay, L.; Bennett, S.N.; Nerurkar, V.R.; Parmenter, C.; Cook, J.A.; Yates, T.L.; Yanagihara, R. Hantavirus in northern short-tailed shrew, United States. Emerg. Infect. Dis. 2007, 13, 1420–1423. [Google Scholar] [CrossRef]
- Song, J.-W.; Kang, H.J.; Song, K.J.; Truong, T.T.; Bennett, S.N.; Arai, S.; Truong, N.U.; Yanagihara, R. Newfound hantavirus in Chinese mole hantavirus in Chinese mole shrew, Vietnam. Emerg. Infect. Dis. 2007, 13, 1784–1787. [Google Scholar] [CrossRef]
- Song, J.-W.; Gu, S.H.; Bennett, S.N.; Arai, S.; Puorger, M.; Hilbe, M.; Yanagihara, R. Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus). Virol. J. 2007, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Bennett, S.N.; Sumibcay, L.; Cook, J.A.; Song, J.-W.; Hope, A.; Parmenter, C.; Nerurkar, V.R.; Yates, T.L.; Yanagihara, R. Phylogenetically distinct hantaviruses in the masked hrew (Sorex cinereus) and dusky shrew (Sorex monticolus) in the United States. Am. J. Trop. Med. Hyg. 2008, 78, 348–351. [Google Scholar]
- Arai, S.; Ohdachi, S.D.; Asakawa, M.; Kang, H.J.; Mocz, G.; Arikawa, J.; Okabe, N.; Yanagihara, R. Molecular phylogeny of a newfound hantavirus in the Japanese shrew mole (Urotrichus talpoides). Proc. Natl. Acad. Sci. USA 2008, 105, 16296–16301. [Google Scholar]
- Song, J.-W.; Kang, H.J.; Gu, S.H.; Moon, S.S.; Bennett, S.N.; Song, K.-J.; Baek, L.J.; Kim, H.C.; O’Guinn, M.L.; Chong, S.T.; et al. Characterization of Imjin virus, a newly isolated hantavirus from the Ussuri white-toothed shrew (Crocidura lasiura). J. Virol. 2009, 83, 6184–6191. [Google Scholar] [CrossRef]
- Kang, H.J.; Bennett, S.N.; Dizney, L.; Sumibcay, L.; Arai, S.; Ruedas, L.A.; Song, J.-W.; Yanagihara, R. Host switch during evolution of a genetically distinct hantavirus in the American shrew mole (Neurotrichus gibbsii). Virology 2009, 388, 8–14. [Google Scholar] [CrossRef]
- Kang, H.J.; Bennett, S.N.; Sumibcay, L.; Arai, S.; Hope, A.G.; Mocz, G.; Song, J.-W.; Cook, J.A.; Yanagihara, R. Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea). PLoS One 2009, 4, e6149. [Google Scholar] [CrossRef]
- Kang, H.J.; Arai, S.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Novel hantavirus in the flat-skulled shrew (Sorex roboratus). Vector Borne Zoonot. Dis. 2010, 10, 593–597. [Google Scholar] [CrossRef]
- Kang, H.J.; Bennett, S.N.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Shared ancestry between a mole-borne hantavirus and hantaviruses harbored by cricetid rodents. J. Virol. 2011, 85, 7496–7503. [Google Scholar] [CrossRef]
- Kang, H.J.; Kadjo, B.; Dubey, S.; Jacquet, F.; Yanagihara, R. Molecular evolution of Azagny virus, a newfound hantavirus harbored by the West African pygmy shrew (Crocidura obscurior) in Côte d’Ivoire. Virol. J. 2011, 8, 373. [Google Scholar] [CrossRef]
- Arai, S.; Gu, S.H.; Baek, L.J.; Tabara, K.; Bennett, S.N.; Oh, H.S.; Takada, N.; Kang, H.J.; Tanaka-Taya, K.; Morikawa, S.; et al. Divergent ancestral lineages of newfound hantaviruses harbored by phylogenetically related crocidurine shrew species in Korea. Virology 2012, 424, 99–105. [Google Scholar] [CrossRef]
- Gu, S.H.; Markowski, J.; Kang, H.J.; Hejduk, J.; Sikorska, B.; Liberski, P.P.; Yanagihara, R. Boginia virus, a newfound hantavirus harbored by the Eurasian water shrew (Neomys fodiens) in Poland. Virol. J. 2013, 10, 160. [Google Scholar] [CrossRef]
- Gu, S.H.; Nicolas, V.; Lalis, A.; Sathirapongsasuti, N.; Yanagihara, R. Complete genome sequence analysis and molecular phylogeny of a newfound hantavirus harbored by the Doucet’s musk shrew (Crocidura douceti) in Guinea. Infect. Genet. Evol. 2013, 20, 118–123. [Google Scholar] [CrossRef]
- Radosa, L.; Schlegel, M.; Gebauer, P.; Ansorge, H.; Heroldová, M.; Jánová, E.; Stanko, M.; Mošanský, L.; Fričová, J.; Pejčoch, M.; et al. Detection of shrew-borne hantavirus in Eurasian pygmy shrew (Sorex minutus) in Central Europe. Infect. Genet. Evol. 2013. [Google Scholar] [CrossRef]
- Mouchaty, S.K.; Gullberg, A.; Janke, A.; Arnason, U. The phylogenetic position of the Talpidae within Eutheria based on analysis of complete mitochondrial sequences. Mol. Biol. Evol. 2000, 17, 60–67. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Penny, D. Implications for bat evolution from two new complete mitochondrial genomes. Mol. Biol. Evol. 2001, 18, 684–688. [Google Scholar] [CrossRef]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Johara, M.; Field, H.; Rashdi, A.; Morrissy, C.; vanderHeide, B.; Rota, P.; Azri, A.; White, J.; Daniels, P.; Jamaluddin, A.; et al. Serological evidence of infection with Nipah virus in bats (order Chiroptera) in Peninsular Malaysia. Emerg. Infect. Dis. 2001, 7, 439–441. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Kim, G.R.; Lee, Y.T.; Park, C.H. A new natural reservoir of hantavirus: Isolation of hantaviruses from lung tissues of bats. Arch. Virol. 1994, 134, 85–95. [Google Scholar] [CrossRef]
- Jung, Y.T.; Kim, G.R. Genomic characterization of M and S RNA segments of hantaviruses isolated from bats. Acta Virol. 1995, 39, 231–233. [Google Scholar]
- Sumibcay, L.; Kadjo, B.; Gu, S.H.; Kang, H.J.; Lim, B.K.; Cook, J.A.; Song, J.-W.; Yanagihara, R. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol. J. 2012, 9, 34. [Google Scholar] [CrossRef]
- Weiss, S.; Witkowski, P.T.; Auste, B.; Nowak, K.; Weber, N.; Fahr, J.; Mombouli, J.V.; Wolfe, N.D.; Drexler, J.F.; Drosten, C.; et al. Hantavirus in bat, Sierra Leone. Emerg. Infect. Dis. 2012, 18, 159–161. [Google Scholar] [CrossRef]
- Arai, S.; Nguyen, S.T.; Boldgiv, B.; Fukui, D.; Araki, K.; Dang, C.N.; Ohdachi, S.D.; Nguyen, N.X.; Pham, T.D.; Boldbaatar, B.; et al. Novel bat-borne hantavirus, Vietnam. Emerg. Infect. Dis. 2013, 19, 1159–1161. [Google Scholar] [CrossRef]
- Guo, W.-P.; Lin, X.-D.; Wang, W.; Tian, J.-H.; Cong, M.-L.; Zhang, H.-L.; Wang, M.-R.; Zhou, R.-H.; Wang, J.-B.; Li, M.-H.; et al. Phylogeny and origins of hantaviruses harbored by bats, insectivores and rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef]
- Maes, P.; Klempa, B.; Clement, J.; Matthijnssens, J.; Gajdusek, D.C.; Krüger, D.H.; van Ranst, M. A proposal for new criteria for the classification of hantaviruses, based on S and M segment protein sequences. Infect. Genet. Evol. 2009, 9, 813–820. [Google Scholar] [CrossRef]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Teeling, E.C. Bats (Chiroptera). In The Timetere of Life; Hedges, S.B., Kumar, S., Eds.; Oxford University Press: Oxford, UK, 2009; pp. 499–503. [Google Scholar]
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.-W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014. [Google Scholar] [CrossRef]
- Bennett, S.N.; Gu, S.H.; Kang, H.J.; Arai, S.; Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014, in press. [Google Scholar]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web-servers. Syst. Biol. 2008, 75, 758–771. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gu, S.H.; Lim, B.K.; Kadjo, B.; Arai, S.; Kim, J.-A.; Nicolas, V.; Lalis, A.; Denys, C.; Cook, J.A.; Dominguez, S.R.; et al. Molecular Phylogeny of Hantaviruses Harbored by Insectivorous Bats in Côte d’Ivoire and Vietnam. Viruses 2014, 6, 1897-1910. https://doi.org/10.3390/v6051897
Gu SH, Lim BK, Kadjo B, Arai S, Kim J-A, Nicolas V, Lalis A, Denys C, Cook JA, Dominguez SR, et al. Molecular Phylogeny of Hantaviruses Harbored by Insectivorous Bats in Côte d’Ivoire and Vietnam. Viruses. 2014; 6(5):1897-1910. https://doi.org/10.3390/v6051897
Chicago/Turabian StyleGu, Se Hun, Burton K. Lim, Blaise Kadjo, Satoru Arai, Jeong-Ah Kim, Violaine Nicolas, Aude Lalis, Christiane Denys, Joseph A. Cook, Samuel R. Dominguez, and et al. 2014. "Molecular Phylogeny of Hantaviruses Harbored by Insectivorous Bats in Côte d’Ivoire and Vietnam" Viruses 6, no. 5: 1897-1910. https://doi.org/10.3390/v6051897