
Increased Viral Dissemination in the Brain and Lethality in MCMV-Infected, Dicer-Deficient Neonates



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
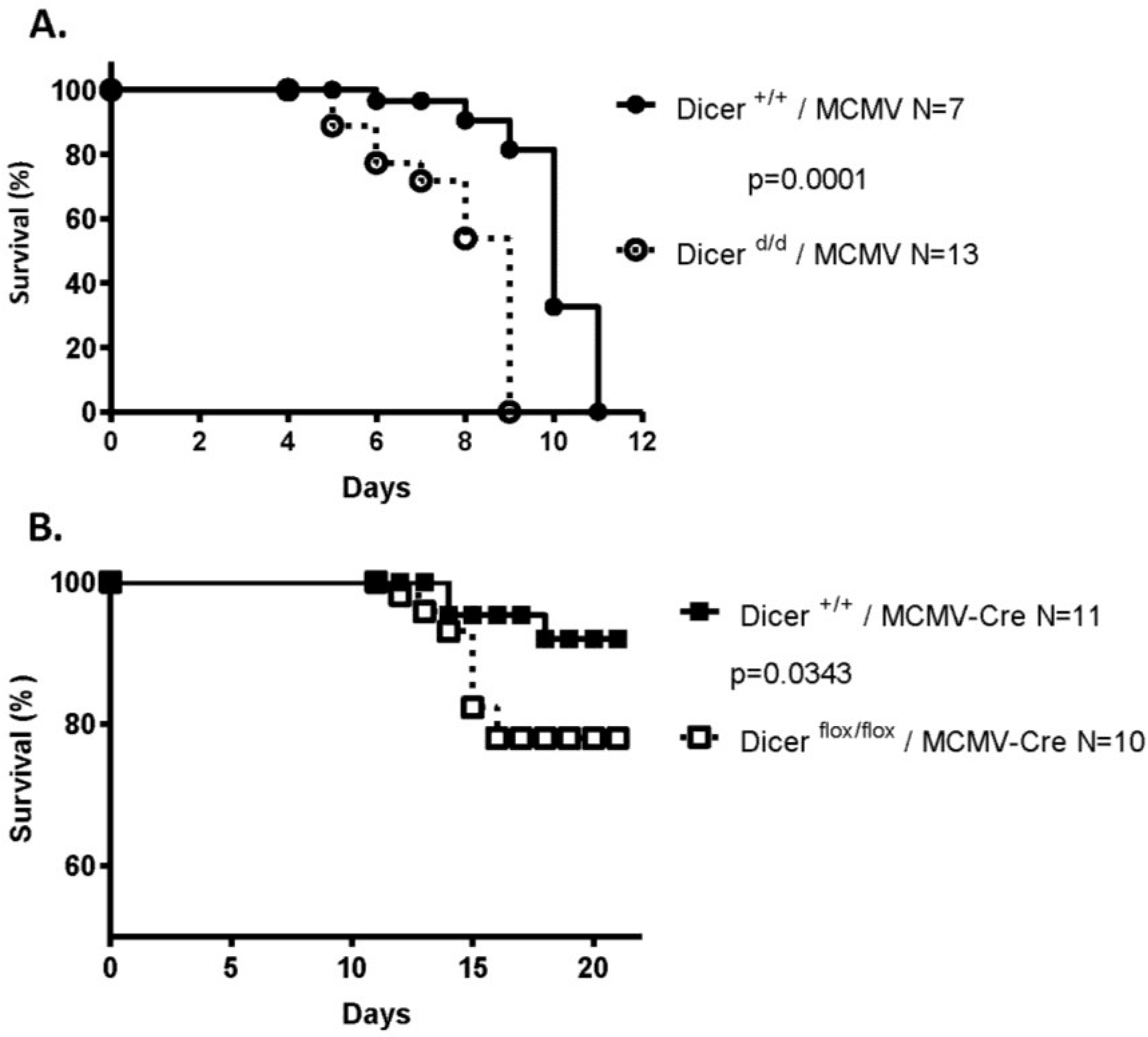
2.1. Impaired miRNA Biogenesis Induces Increased Lethality in MCMV-Infected Mouse Neonates

2.2. Increased Viral Replication in Dicer-Deficient Newborns

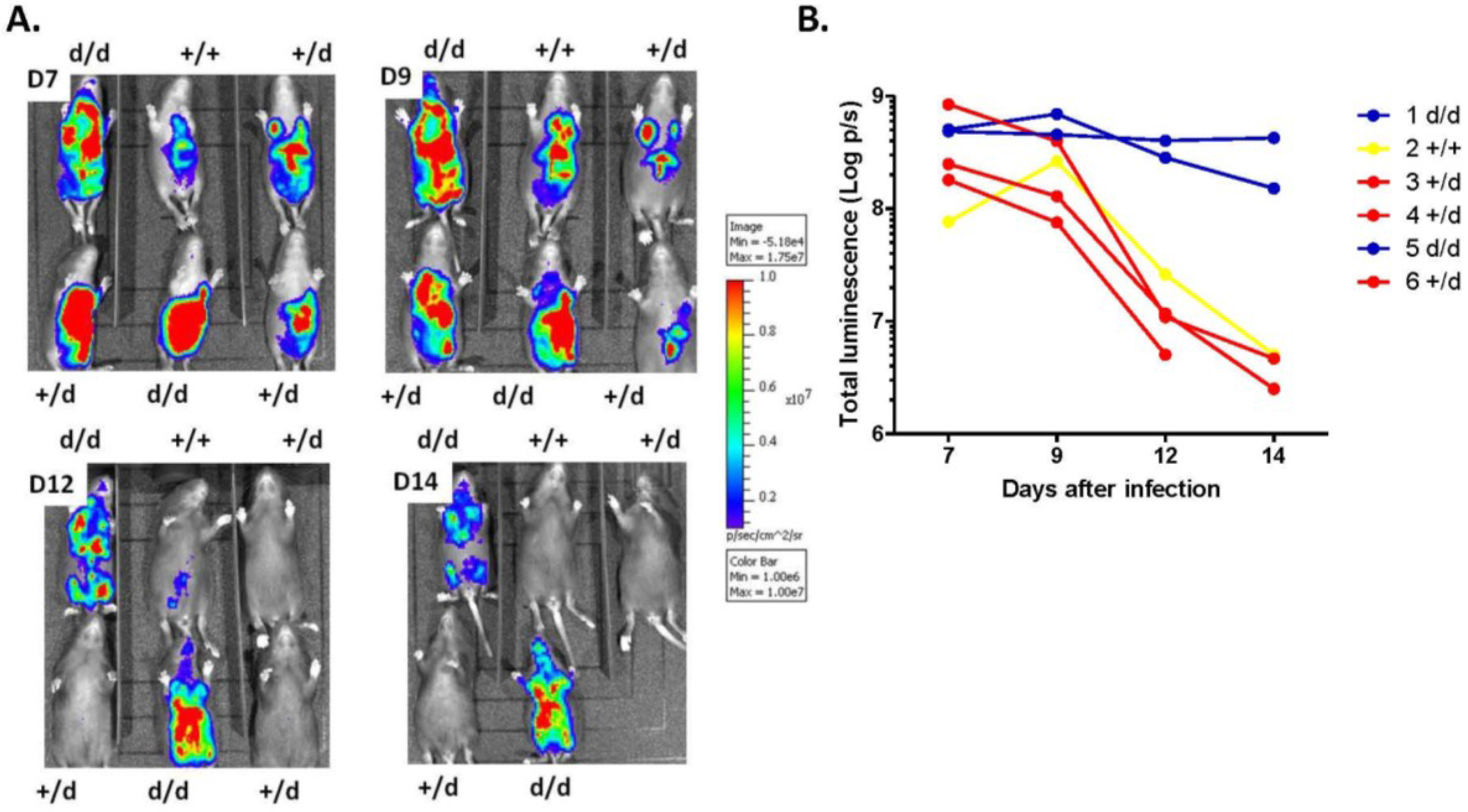
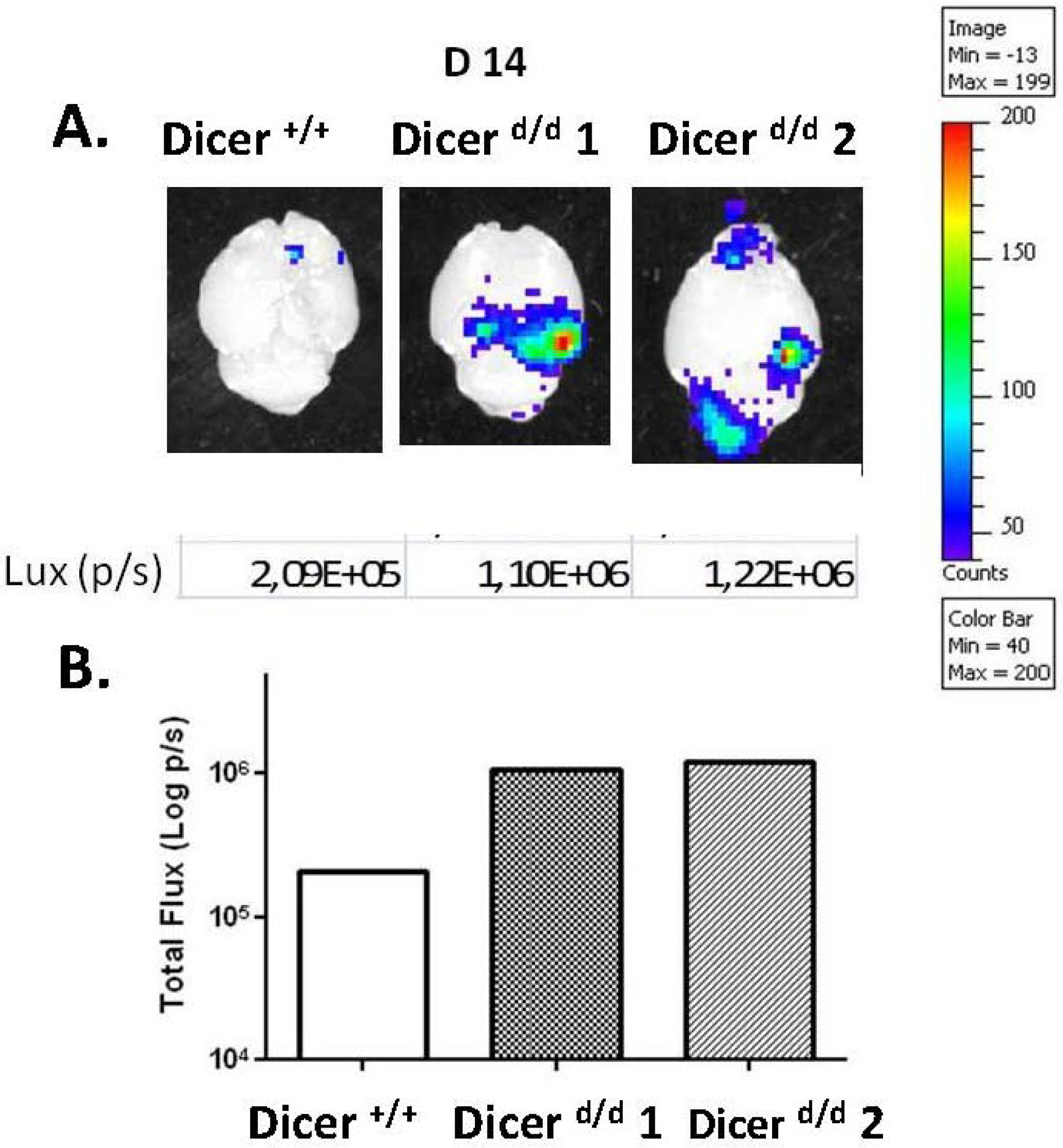
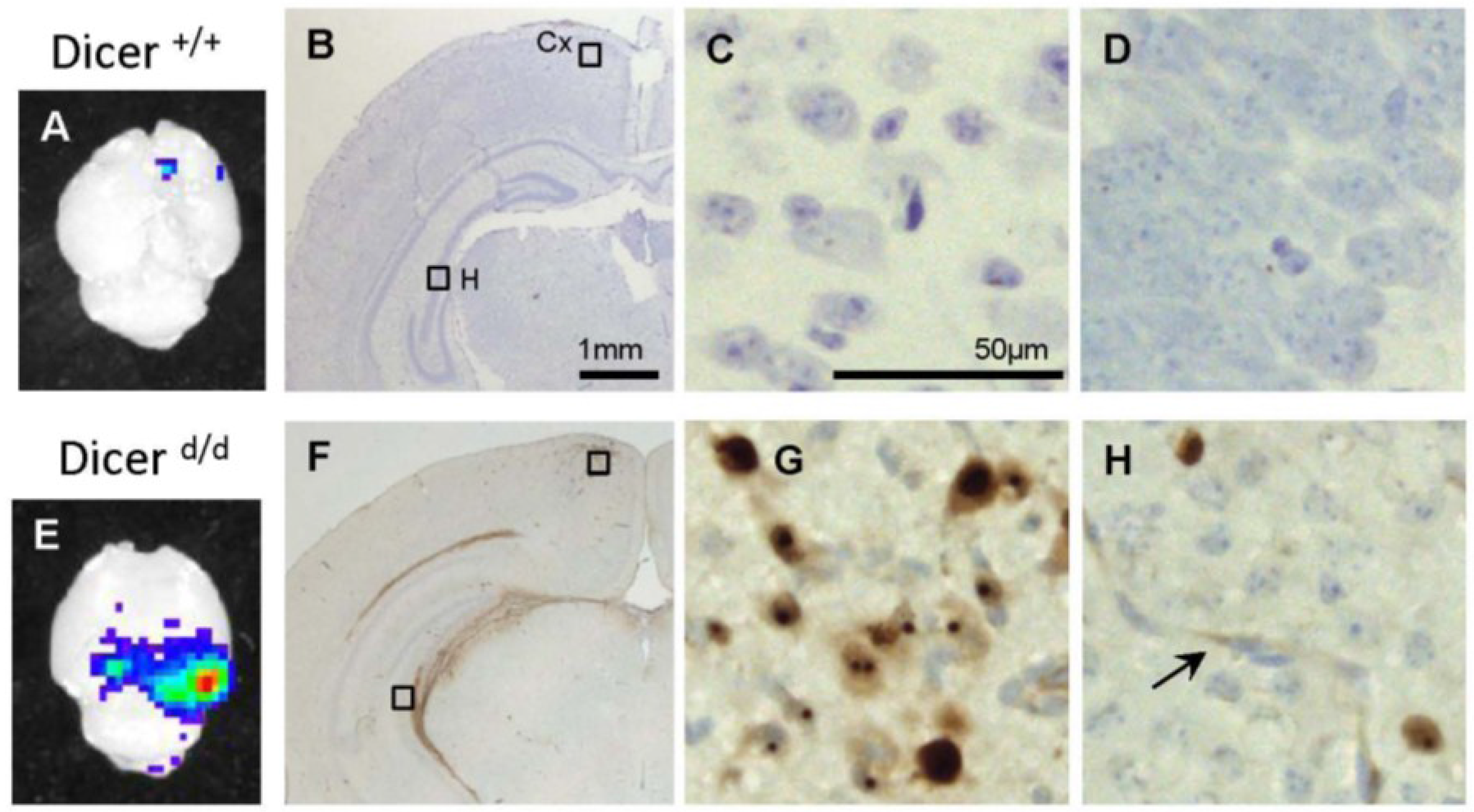
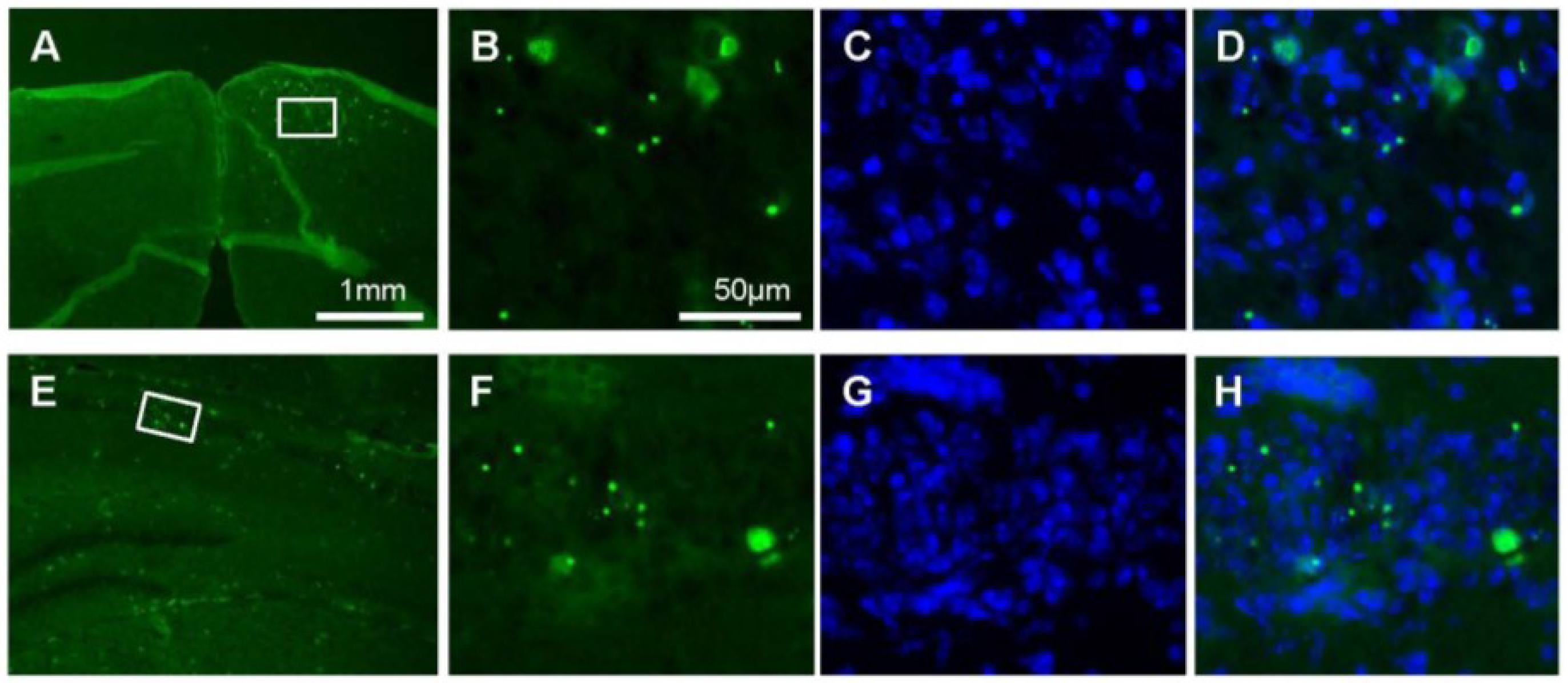
2.3. Preferential Viral Dissemination in the Brain of Dicer-Deficient Newborns




3. Discussion
4. Materials and Methods
4.1. Mice and Ethics Statement
4.2. Viruses
4.3. In Vivo Imaging
4.4. Immunohistochemistry Staining
4.5. Statistical Analysis
Supplementary Files
Supplementary File 1Acknowledgment
Author Contributions
Conflicts of Interest
References
- Loewendorf, A.; Benedict, C.A. Modulation of host innate and adaptive immune defenses by cytomegalovirus: Timing is everything. J. Intern. Med. 2010, 267, 483–501. [Google Scholar] [CrossRef]
- Lischka, P.; Zimmermann, H. Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Curr. Opin. Pharmacol. 2008, 8, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Baryawno, N.; Soderberg-Naucler, C. Is human cytomegalovirus a target in cancer therapy? Oncotarget 2011, 2, 1329–1338. [Google Scholar] [PubMed]
- Morein, B.; Abusugra, I.; Blomqvist, G. Immunity in neonates. Vet. Immunol. Immunopathol. 2002, 87, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Zaghouani, H.; Hoeman, C.M.; Adkins, B. Neonatal immunity: Faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol. 2009, 30, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Morein, B.; Blomqvist, G.; Hu, K. Immune responsiveness in the neonatal period. J. Comp. Pathol. 2007, 137, S27–S31. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, M.C.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of congenital cytomegalovirus infection: Disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 2009, 22, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, Y. Effects of cytomegalovirus infection on embryogenesis and brain development. Congenit. Anom. 2009, 49, 47–55. [Google Scholar] [CrossRef]
- Sancho-Shimizu, V.; Perez de Diego, R.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Invest. 2011, 121, 4889–4902. [Google Scholar] [CrossRef] [PubMed]
- Renneson, J.; Dutta, B.; Goriely, S.; Danis, B.; Lecomte, S.; Laes, J.F.; Tabi, Z.; Goldman, M.; Marchant, A. IL-12 and type I IFN response of neonatal myeloid DC to human CMV infection. Eur. J. Immunol. 2009, 39, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Shimizu, V.; Zhang, S.Y.; Abel, L.; Tardieu, M.; Rozenberg, F.; Jouanguy, E.; Casanova, J.L. Genetic susceptibility to herpes simplex virus 1 encephalitis in mice and humans. Curr. Opin. Allergy Clin. Immunol. 2007, 7, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Crozat, K.; Georgel, P.; Rutschmann, S.; Mann, N.; Du, X.; Hoebe, K.; Beutler, B. Analysis of the MCMV resistome by ENU mutagenesis. Mamm. Genome 2006, 17, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Tabeta, K.; Georgel, P.; Janssen, E.; Du, X.; Hoebe, K.; Crozat, K.; Mudd, S.; Shamel, L.; Sovath, S.; Goode, J.; et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3516–3521. [Google Scholar] [CrossRef]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.; Dzionek, A.; Pingel, J.T.; Orihuela, M.M.; Akira, S.; Yokoyama, W.M.; Colonna, M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004, 21, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Georgel, P.; Rutschmann, S.; Jiang, Z.; Croker, B.; Crozat, K. Genetic analysis of innate resistance to mouse cytomegalovirus (MCMV). Brief Funct. Genomic. Proteomic. 2005, 4, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Juanjuan, C.; Yan, F.; Li, C.; Haizhi, L.; Ling, W.; Xinrong, W.; Juan, X.; Tao, L.; Zongzhi, Y.; Suhua, C. Murine model for congenital CMV infection and hearing impairment. Virol. J. 2011, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Koontz, T.; Bralic, M.; Tomac, J.; Pernjak-Pugel, E.; Bantug, G.; Jonjic, S.; Britt, W.J. Altered development of the brain after focal herpesvirus infection of the central nervous system. J. Exp. Med. 2008, 205, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Bantug, G.R.; Cekinovic, D.; Bradford, R.; Koontz, T.; Jonjic, S.; Britt, W.J. CD8+ T lymphocytes control murine cytomegalovirus replication in the central nervous system of newborn animals. J. Immunol. 2008, 181, 2111–2123. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Macquin, C.; Bahram, S.; Georgel, P. Use of in vivo imaging to monitor the progression of experimental mouse cytomegalovirus infection in neonates. J. Vis. Exp. 2013. [Google Scholar] [CrossRef]
- Wells, D.J. Animal welfare and the 3Rs in European biomedical research. Ann. NY Acad. Sci. 2011, 1245, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Rana, T.M. RNA-based mechanisms regulating host-virus interactions. Immunol. Rev. 2013, 253, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Dolken, L.; Pfeffer, S.; Koszinowski, U.H. Cytomegalovirus microRNAs. Virus Genes 2009, 38, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Tuddenham, L.; Macquin, C.; Alsaleh, G.; Schreiber-Becker, J.; Tanguy, M.; Bahram, S.; Pfeffer, S.; Georgel, P. Deregulation of type I IFN-dependent genes correlates with increased susceptibility to cytomegalovirus acute infection of dicer mutant mice. PLOS ONE 2012, 7, e43744. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Jing, Q.; Georgel, P.; New, L.; Chen, J.; Mols, J.; Kang, Y.J.; Jiang, Z.; Du, X.; Cook, R.; et al. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 2007, 27, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Mutnal, M.B.; Cheeran, M.C.; Hu, S.; Lokensgard, J.R. Murine cytomegalovirus infection of neural stem cells alters neurogenesis in the developing brain. PLOS ONE 2011, 6, e16211. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, A.; Halle, S.; Seckert, C.K.; Lemmermann, N.A.; Veres, T.Z.; Braun, A.; Maus, U.A.; Forster, R.; Reddehase, M.J.; Messerle, M.; et al. Single cell detection of latent cytomegalovirus reactivation in host tissue. J. Gen. Virol. 2011, 92, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Cekinovic, D.; Golemac, M.; Pugel, E.P.; Tomac, J.; Cicin-Sain, L.; Slavuljica, I.; Bradford, R.; Misch, S.; Winkler, T.H.; Mach, M.; et al. Passive immunization reduces murine cytomegalovirus-induced brain pathology in newborn mice. J. Virol. 2008, 82, 12172–12180. [Google Scholar] [CrossRef] [PubMed]
- MacKay, C.R.; Wang, J.P.; Kurt-Jones, E.A. Dicer’s role as an antiviral: Still an enigma. Curr. Opin. Immunol. 2014, 26, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Van Stry, M.; Oguin, T.H., 3rd; Cheloufi, S.; Vogel, P.; Watanabe, M.; Pillai, M.R.; Dash, P.; Thomas, P.G.; Hannon, G.J.; Bix, M. Enhanced susceptibility of Ago1/3 double-null mice to influenza A virus infection. J. Virol. 2012, 86, 4151–4157. [Google Scholar]
- Sacher, T.; Andrassy, J.; Kalnins, A.; Dolken, L.; Jordan, S.; Podlech, J.; Ruzsics, Z.; Jauch, K.W.; Reddehase, M.J.; Koszinowski, U.H. Shedding light on the elusive role of endothelial cells in cytomegalovirus dissemination. PLOS Pathog. 2011, 7, e1002366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Olson, E.N. AngiomiRs—Key regulators of angiogenesis. Curr. Opin. Genet. Dev. 2009, 19, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Murchison, E.P.; Partridge, J.F.; Tam, O.H.; Cheloufi, S.; Hannon, G.J. Characterization of Dicer-deficient murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 2005, 102, 12135–12140. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; Wang, B.; Terhorst, C.; Biron, C.A. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 1995, 182, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Cicin-Sain, L.; Podlech, J.; Messerle, M.; Reddehase, M.J.; Koszinowski, U.H. Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J. Virol. 2005, 79, 9492–9502. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostermann, E.; Macquin, C.; Krezel, W.; Bahram, S.; Georgel, P. Increased Viral Dissemination in the Brain and Lethality in MCMV-Infected, Dicer-Deficient Neonates. Viruses 2015, 7, 2308-2320. https://doi.org/10.3390/v7052308
Ostermann E, Macquin C, Krezel W, Bahram S, Georgel P. Increased Viral Dissemination in the Brain and Lethality in MCMV-Infected, Dicer-Deficient Neonates. Viruses. 2015; 7(5):2308-2320. https://doi.org/10.3390/v7052308
Chicago/Turabian StyleOstermann, Eleonore, Cécile Macquin, Wojciech Krezel, Seiamak Bahram, and Philippe Georgel. 2015. "Increased Viral Dissemination in the Brain and Lethality in MCMV-Infected, Dicer-Deficient Neonates" Viruses 7, no. 5: 2308-2320. https://doi.org/10.3390/v7052308
APA StyleOstermann, E., Macquin, C., Krezel, W., Bahram, S., & Georgel, P. (2015). Increased Viral Dissemination in the Brain and Lethality in MCMV-Infected, Dicer-Deficient Neonates. Viruses, 7(5), 2308-2320. https://doi.org/10.3390/v7052308