
Autophagy Activated by Bluetongue Virus Infection Plays a Positive Role in Its Replication

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses and Plasmids
2.2. Virus Infection and Titration Assays
2.3. SDS-PAGE and Western Blot
2.4. Transmission Electron Microscopy
2.5. Confocal Fluorescence Microscopy
2.6. Preparation of Ultraviolet (UV)-Inactivated BTV1
2.7. Cell Viability and Drug Treatment Assay
2.8. RNA Interference of Beclin1
2.9. Statistical Analysis
3. Results
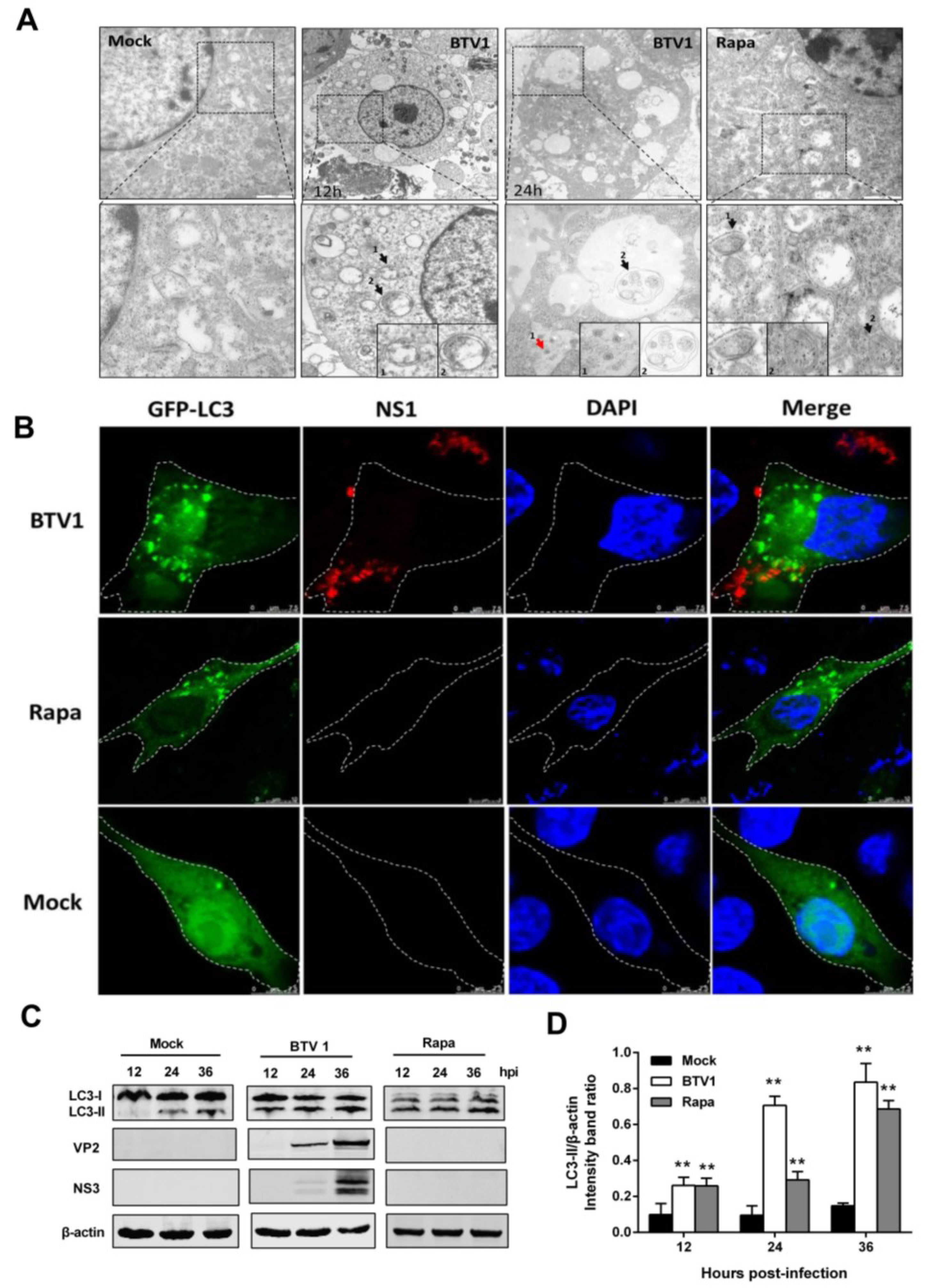
3.1. Notable Autophagy is Triggered by BTV1 Infection in BSR Cells

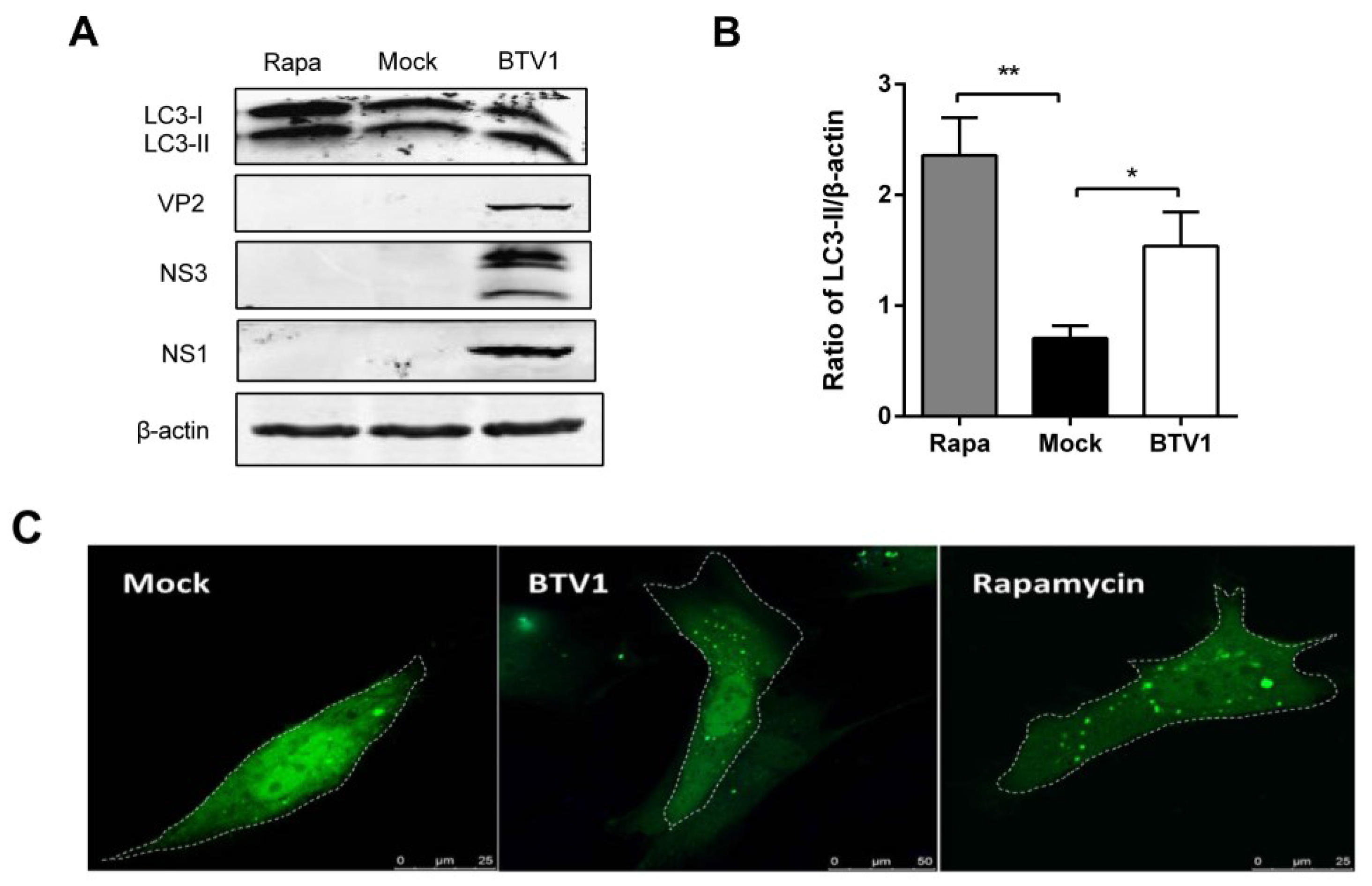
3.2. Autophagy is Induced in Primary Lamb Lingual Epithelial Cells by BTV1 Infection
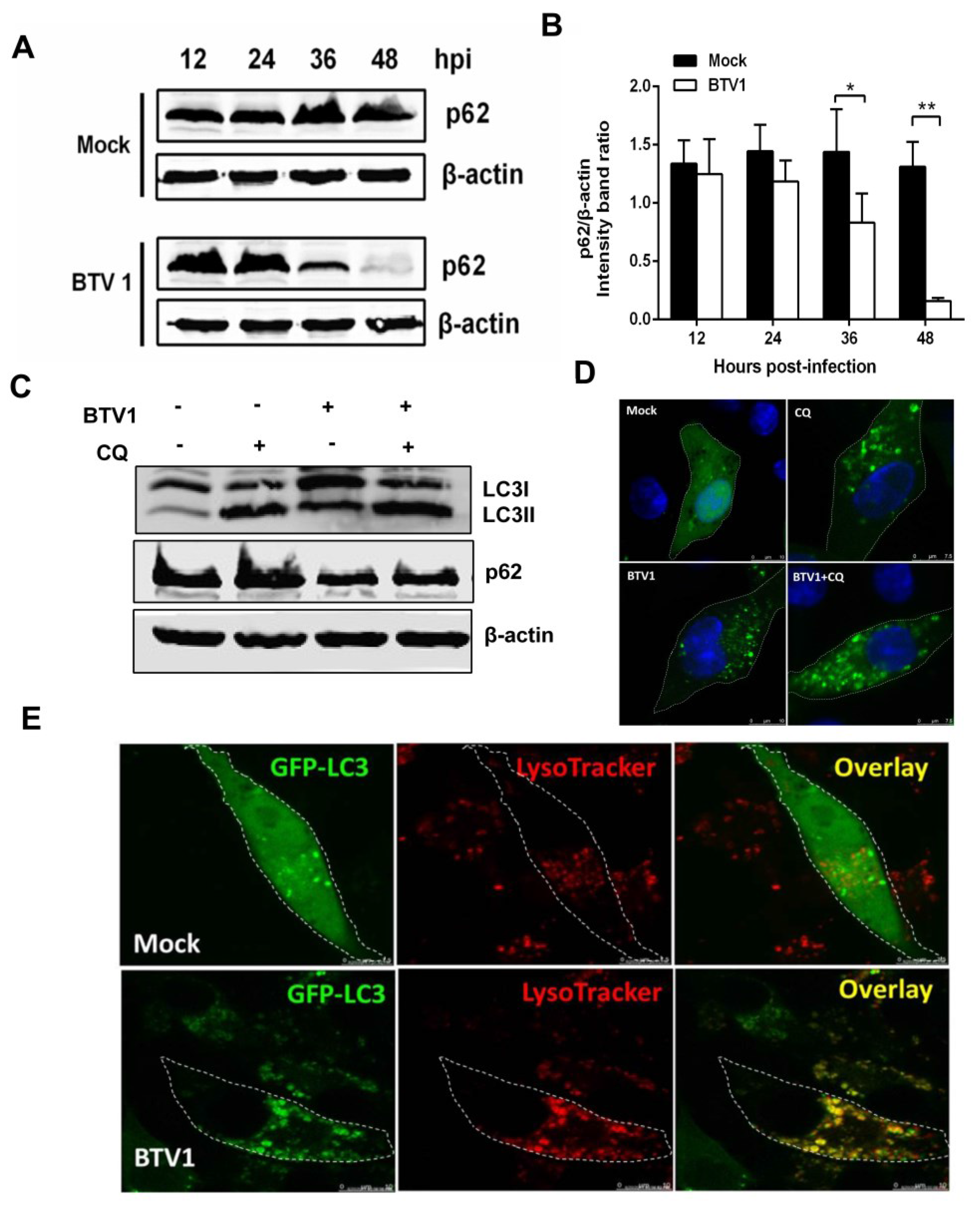
3.3. BTV1 Infection Increases the Levels of Autophagic Flux


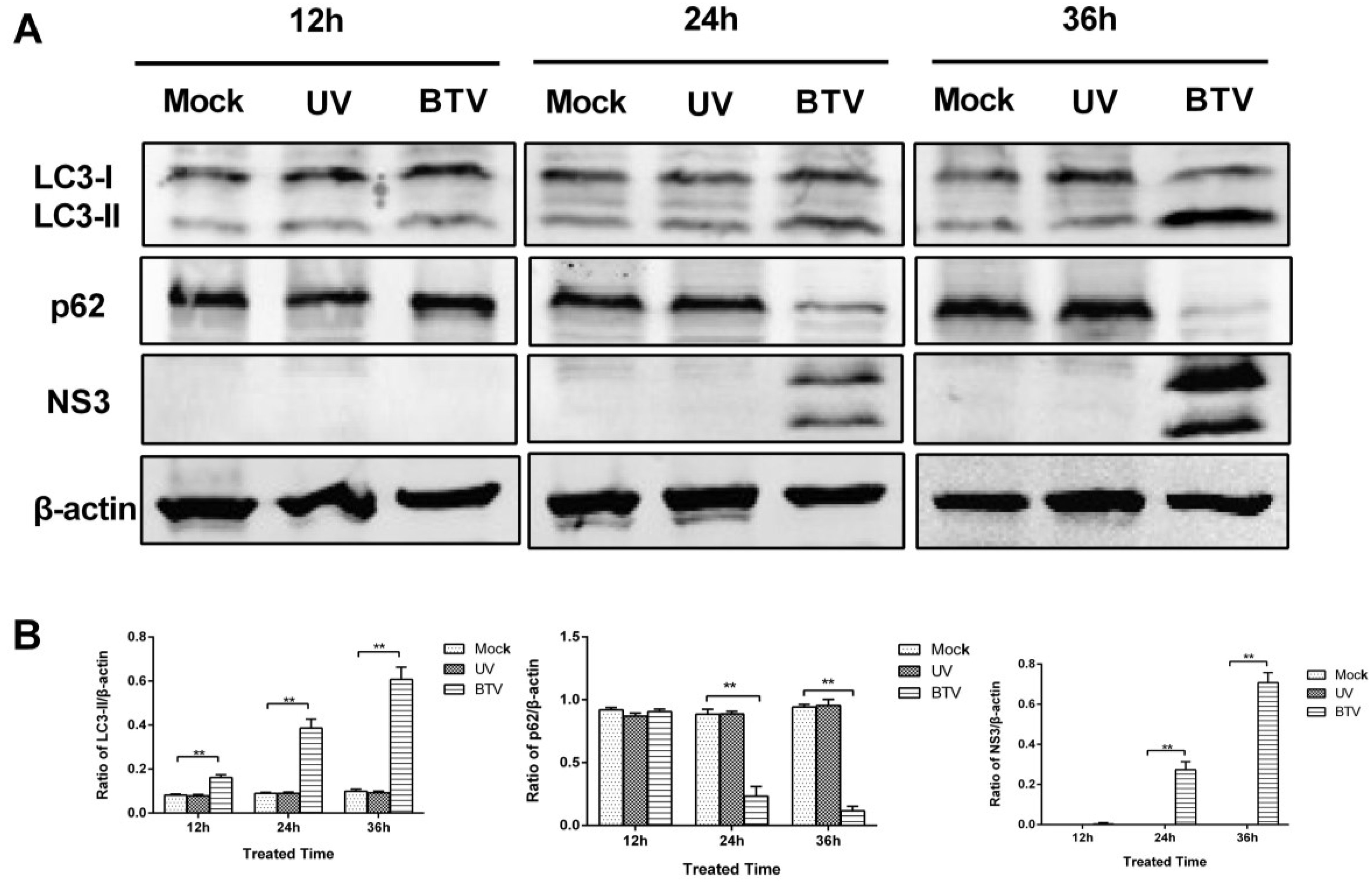
3.4. The Autophagy is Dependent on BTV-Productive Infection

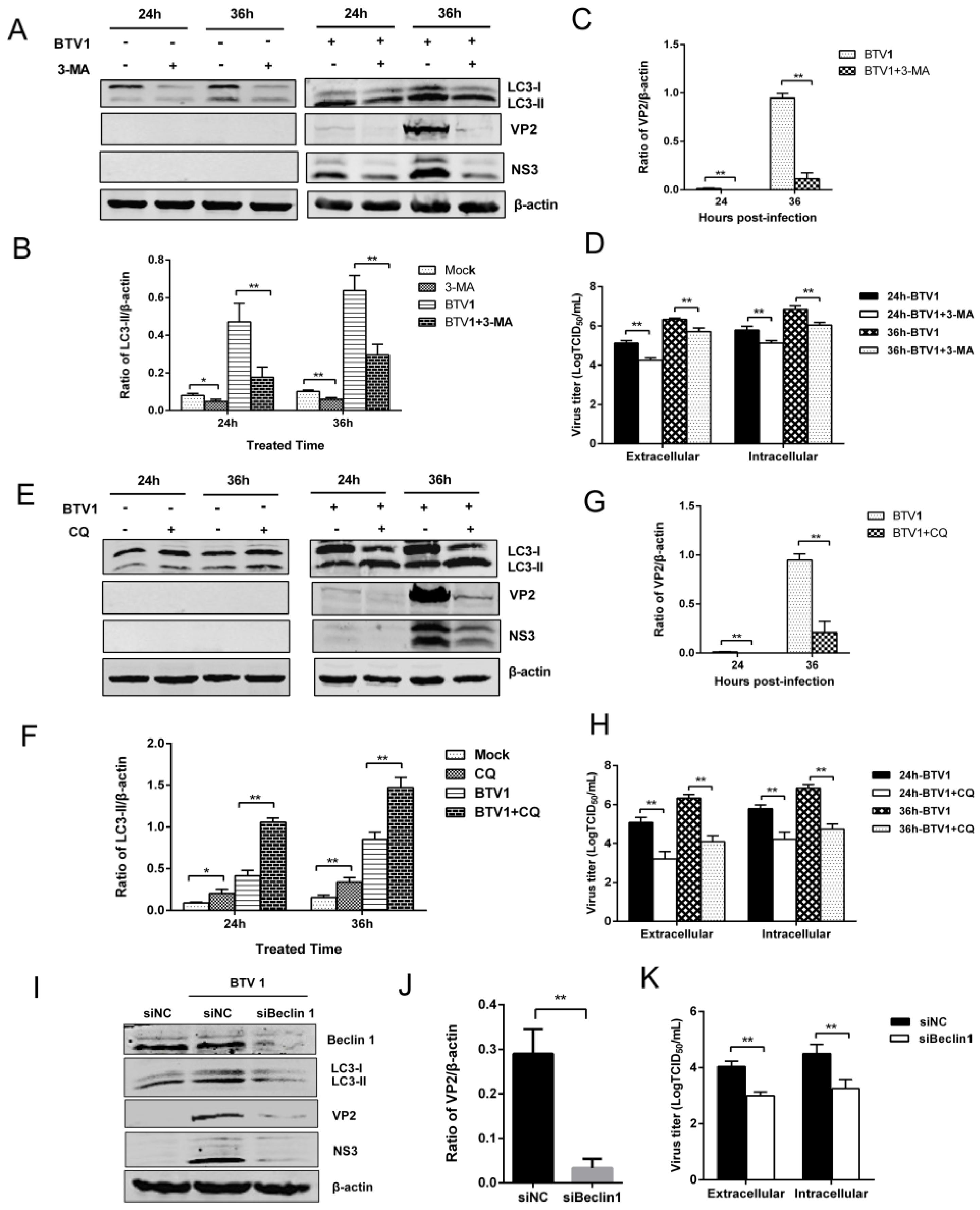
3.5. Inhibition of Autophagy Reduced BTV1 Replication

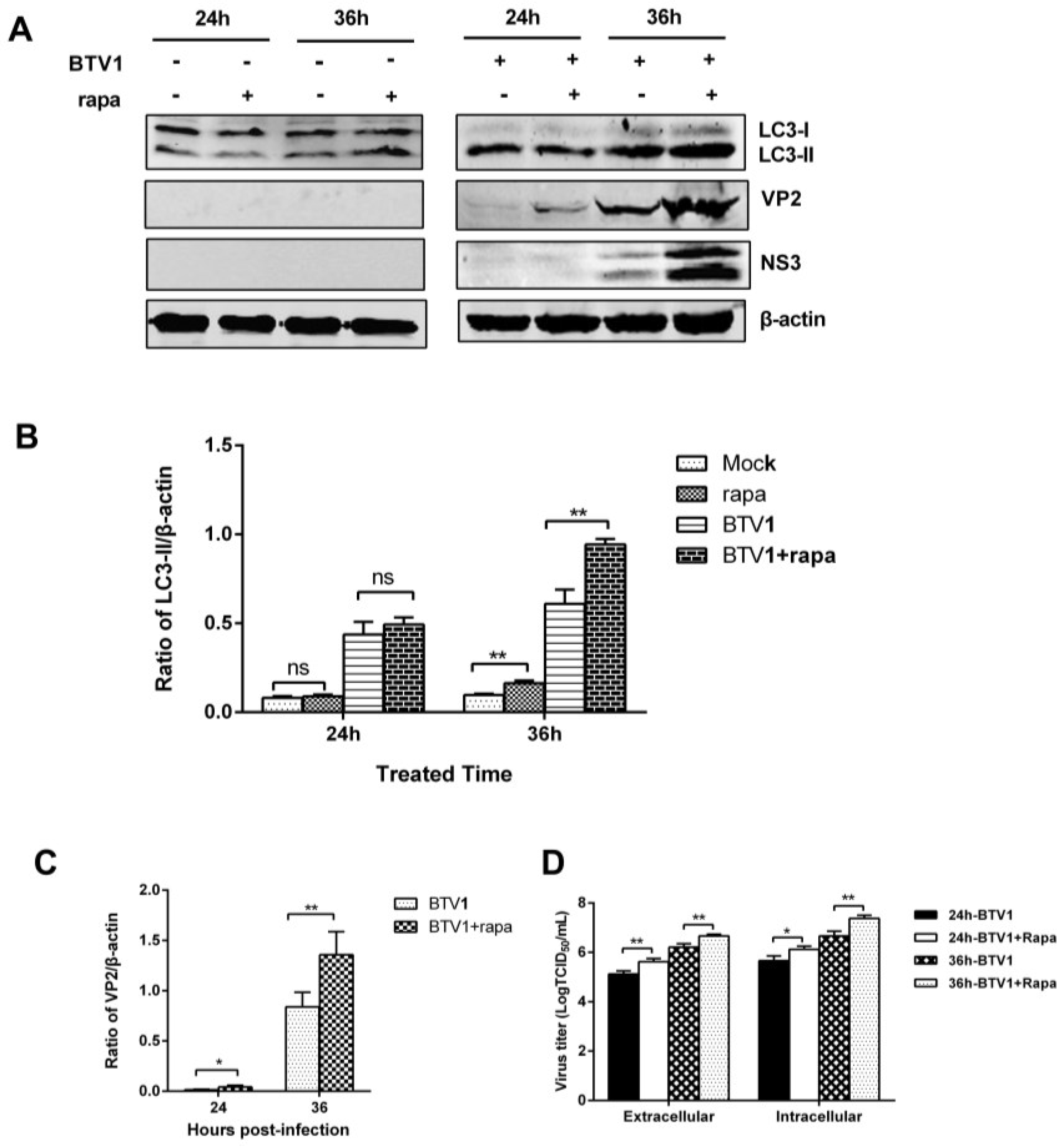
3.6. Induction of Autophagy Promotes BTV1 Replication

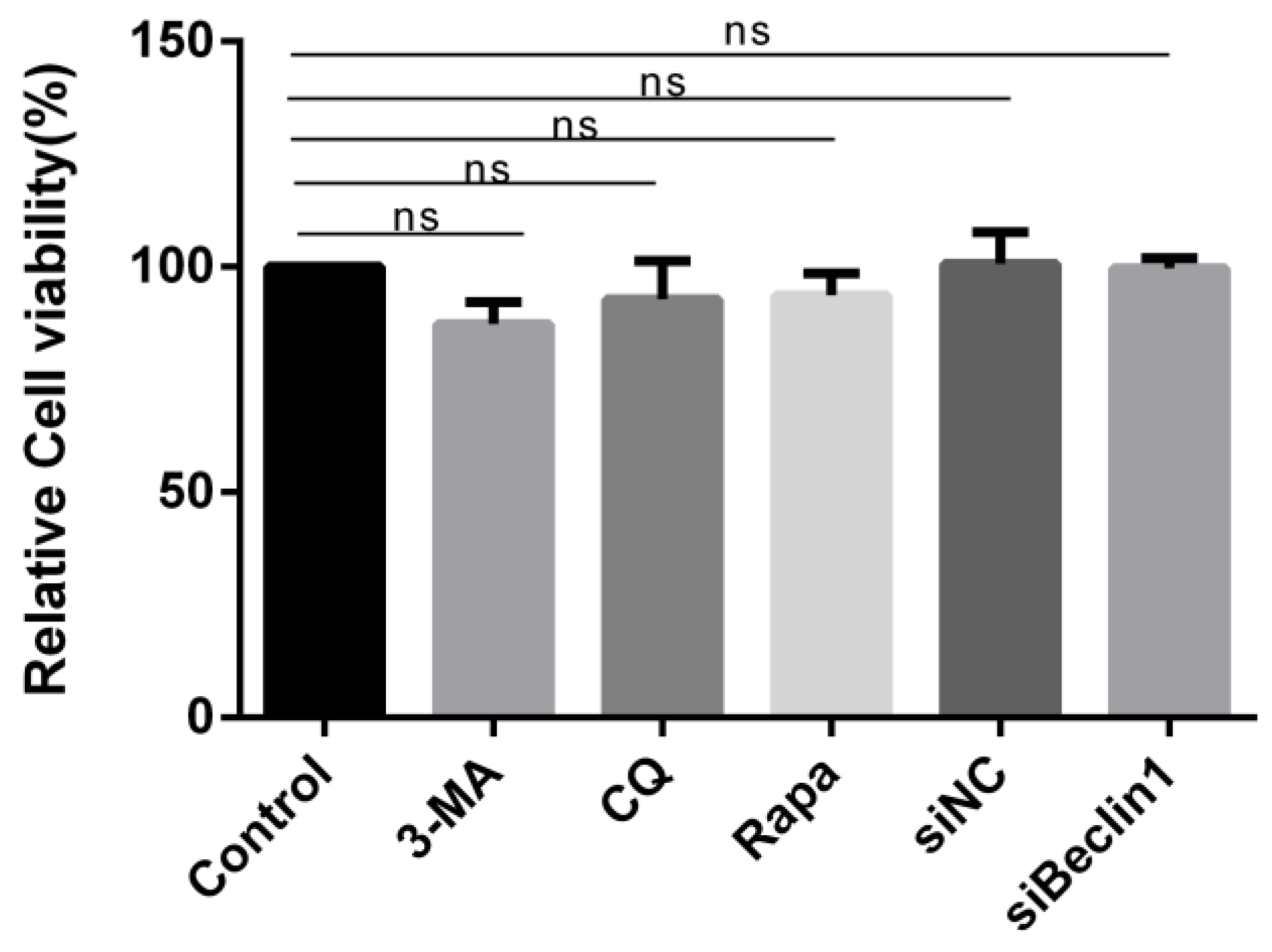
3.7. Cell Viability Unaffected by Pharmacological Treatment

4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maclachlan, N.J. Bluetongue: History, global epidemiology, and pathogenesis. Prev. Vet. Med. 2011, 102, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Mertens, P.P.; Diprose, J.; Maan, S.; Singh, K.P.; Attoui, H.; Samuel, A.R. Bluetongue virus replication, molecular and structural biology. Vet. Ital. 2004, 40, 426–437. [Google Scholar] [PubMed]
- Schwartz-Cornil, I.; Mertens, P.P.; Contreras, V.; Hemati, B.; Pascale, F.; Breard, E.; Mellor, P.S.; MacLachlan, N.J.; Zientara, S. Bluetongue virus: Virology, pathogenesis and immunity. Vet. Res. 2008, 39, e46. [Google Scholar] [CrossRef] [PubMed]
- Roy, P. Bluetongue virus proteins and particles and their role in virus entry, assembly, and release. Adv. Virus Res. 2005, 64, 69–123. [Google Scholar] [PubMed]
- Roy, P. Functional mapping of bluetongue virus proteins and their interactions with host proteins during virus replication. Cell Biochem. Biophys. 2008, 50, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Roy, P. Bluetongue virus: Dissection of the polymerase complex. J. Gen. Virol. 2008, 89, 1789–1804. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Roy, P. The molecular biology of bluetongue virus replication. Virus Res. 2014, 182, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Dawson, P.W.H.; Richardson, C.D. Viral interactions with macroautophagy: A double-edged sword. Virology 2010, 402, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Piacentini, M. Toward the understanding of autophagy regulation and its interplay with cell death pathways. Cell Death Differ. 2009, 16, 933–934. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.C.; Richart, S.; Lin, F.Y.; Hsu, W.L.; Liu, H.J. The interplay of reovirus with autophagy. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy plays an essential anti-viral role in drosophila against vesicular stomatitis virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.X.; Randall, G. Manipulation or capitulation: Virus interactions with autophagy. Microbes Infect. 2012, 14, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.; Jung, J. When autophagy meets viruses: A double-edged sword with functions in defense and offense. Semin. Immunopathol. 2010, 32, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza a virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Musiienko, V.; Bai, Z.; Qin, A.; Schneller, S.W.; Li, Q. Novel virostatic agents against bluetongue virus. PLoS ONE 2012, 7, e43341. [Google Scholar] [CrossRef] [PubMed]
- Shai, B.; Schmukler, E.; Yaniv, R.; Ziv, N.; Horn, G.; Bumbarov, V.; Yadin, H.; Smorodinsky, N.I.; Bacharach, E.; Pinkas-Kramarski, R.; et al. Epizootic hemorrhagic disease virus induces and benefits from cell stress, autophagy, and apoptosis. J. Virol. 2013, 87, 13397–13408. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy 2013, 9, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Jiang, K.; Zhang, X.; Zhang, M.; Zhou, Z.; Hu, M.; Yang, R.; Sun, C.; Wu, Y. Avian reovirus triggers autophagy in primary chicken fibroblast cells and vero cells to promote virus production. Arch. Virol. 2012, 157, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. (Warsz.) 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- DeMaula, C.D.; Jutila, M.A.; Wilson, D.W.; MacLachlan, N.J. Infection kinetics, prostacyclin release and cytokine-mediated modulation of the mechanism of cell death during bluetongue virus infection of cultured ovine and bovine pulmonary artery and lung microvascular endothelial cells. J. Gen. Virol. 2001, 82, 787–794. [Google Scholar] [PubMed]
- Mortola, E.; Noad, R.; Roy, P. Bluetongue virus outer capsid proteins are sufficient to trigger apoptosis in mammalian cells. J. Virol. 2004, 78, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.E.; Ward, S.L.; Mizushima, N.; Levine, B.; Leib, D.A. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 2007, 81, 12128–12134. [Google Scholar] [CrossRef] [PubMed]
- Ruscanu, S.; Pascale, F.; Bourge, M.; Hemati, B.; Elhmouzi-Younes, J.; Urien, C.; Bonneau, M.; Takamatsu, H.; Hope, J.; Mertens, P.; et al. The double-stranded rna bluetongue virus induces type i interferon in plasmacytoid dendritic cells via a myd88-dependent tlr7/8-independent signaling pathway. J. Virol. 2012, 86, 5817–5828. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. P62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Neufeld, T.P. Autophagy: A forty-year search for a missing membrane source. PLoS Biol. 2006, 4, e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannagé, M.; Schmid, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza a virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.C.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in ht-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Meijer, A.J.; Codogno, P. Autophagy and p70s6 kinase. Autophagy 2005, 1, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Ou, J.H.J. Autophagy in viral replication and pathogenesis. Mol. Cells 2010, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimorim, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.B.; Ou, J.H.J. Induction of incomplete autophagic response by hepatitis c virus via the unfolded protein response. Hepatol. (Baltim. Md.) 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.X.; Huang, L.; Wang, R.; Yu, Y.L.; Li, C.; Li, P.P.; Hu, X.C.; Hao, H.P.; Ishag, H.A.; Mao, X. Porcine reproductive and respiratory syndrome virus induces autophagy to promote virus replication. Autophagy 2012, 8, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Jain, B.; Chaturvedi, U.C.; Jain, A. Role of intracellular events in the pathogenesis of dengue; an overview. Microb. Pathog. 2014, 69–70, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Hou, W.H.; Liu, I.H.; Hsiao, G.; Huang, S.S.; Huang, J.S. Inhibitors of clathrin-dependent endocytosis enhance tgfbeta signaling and responses. J. Cell Sci. 2009, 122, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]
- Barhoom, S.; Kaur, J.; Cooperman, B.S.; Smorodinsky, N.I.; Smilansky, Z.; Ehrlich, M.; Elroy-Stein, O. Quantitative single cell monitoring of protein synthesis at subcellular resolution using fluorescently labeled trna. Nucleic Acids Res. 2011, 39, e129. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Jackson, W.T. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 2012, 8, e1003046. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis c virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.T.; Hyatt, A.D.; Brookes, S.M. The replication of bluetongue virus. Curr. Top. Microbiol. Immunol. 1990, 162, 89–118. [Google Scholar] [PubMed]
- Hassan, S.H.; Wirblich, C.; Forzan, M.; Roy, P. Expression and functional characterization of bluetongue virus vp5 protein: Role in cellular permeabilization. J. Virol. 2001, 75, 8356–8367. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, S.; Xu, Q.; Sun, E.; Yang, T.; Li, J.; Feng, Y.; Zhang, Q.; Wang, H.; Zhang, J.; Wu, D. Autophagy Activated by Bluetongue Virus Infection Plays a Positive Role in Its Replication. Viruses 2015, 7, 4657-4675. https://doi.org/10.3390/v7082838
Lv S, Xu Q, Sun E, Yang T, Li J, Feng Y, Zhang Q, Wang H, Zhang J, Wu D. Autophagy Activated by Bluetongue Virus Infection Plays a Positive Role in Its Replication. Viruses. 2015; 7(8):4657-4675. https://doi.org/10.3390/v7082838
Chicago/Turabian StyleLv, Shuang, Qingyuan Xu, Encheng Sun, Tao Yang, Junping Li, Yufei Feng, Qin Zhang, Haixiu Wang, Jikai Zhang, and Donglai Wu. 2015. "Autophagy Activated by Bluetongue Virus Infection Plays a Positive Role in Its Replication" Viruses 7, no. 8: 4657-4675. https://doi.org/10.3390/v7082838
APA StyleLv, S., Xu, Q., Sun, E., Yang, T., Li, J., Feng, Y., Zhang, Q., Wang, H., Zhang, J., & Wu, D. (2015). Autophagy Activated by Bluetongue Virus Infection Plays a Positive Role in Its Replication. Viruses, 7(8), 4657-4675. https://doi.org/10.3390/v7082838