
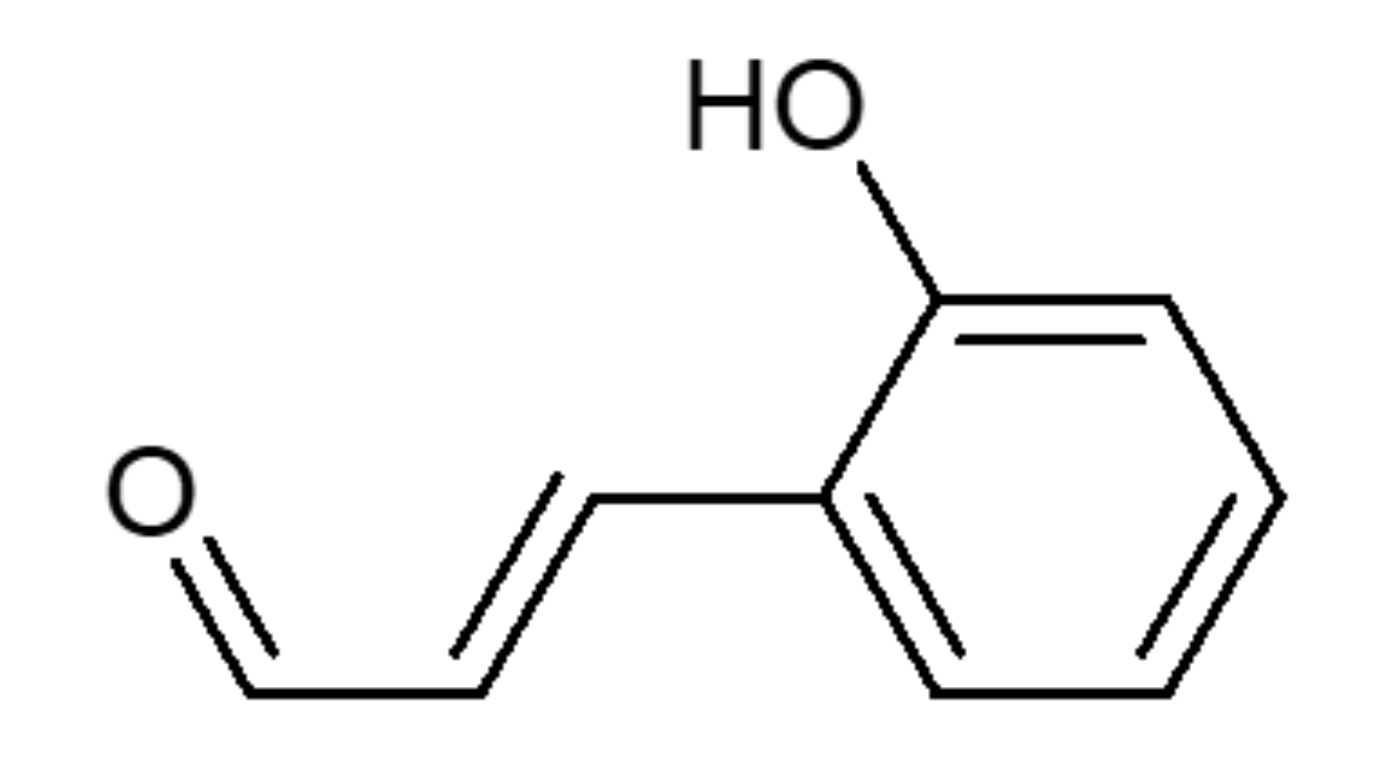
Regulation of ROS-Dependent JNK Pathway by 2’-Hydroxycinnamaldehyde Inducing Apoptosis in Human Promyelocytic HL-60 Leukemia Cells

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Cell Culture
2.2. MTT Assay
2.3. Annexin V-FITC/Propidium Iodide (PI) Double Staining Analysis
2.4. Detection of DNA Fragmentation
2.5. Preparation of Mitochondrial and Cytosolic Fraction and Total Protein
2.6. Western Blot Analysis
2.7. Determination of Mitochondrial Membrane Potential (∆Ψm)
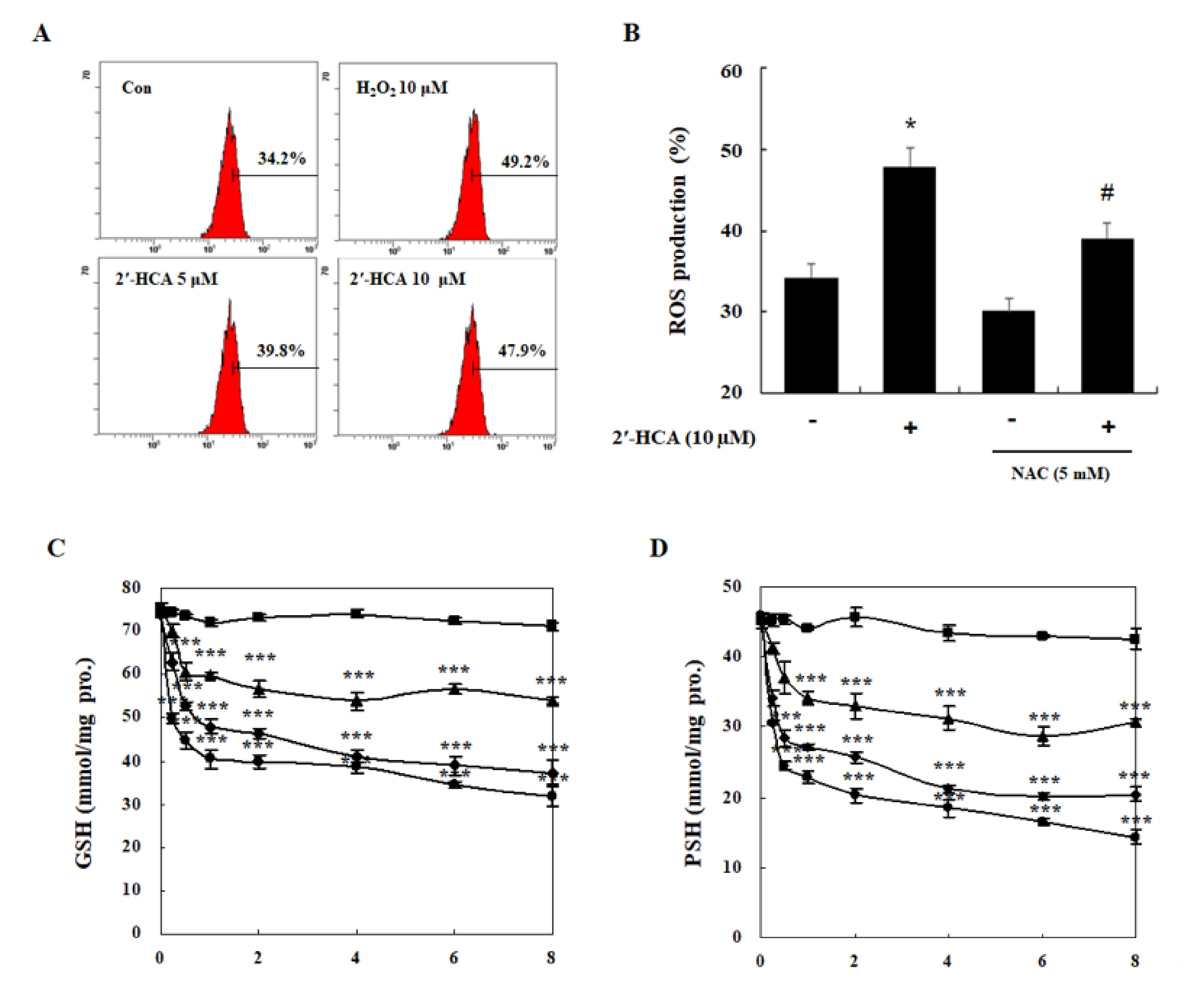
2.8. Detection of ROS Generation
2.9. Determination of the GSH Level
2.10. Measurement of Intracellular Protein Thiols (PSH)
2.11. Caspase-3 Activity Assay
2.12. Transfection for RNA Interference
2.13. JNK Kinase Assay
2.14. Electrophoretic Mobility Shift Assay (EMSA)
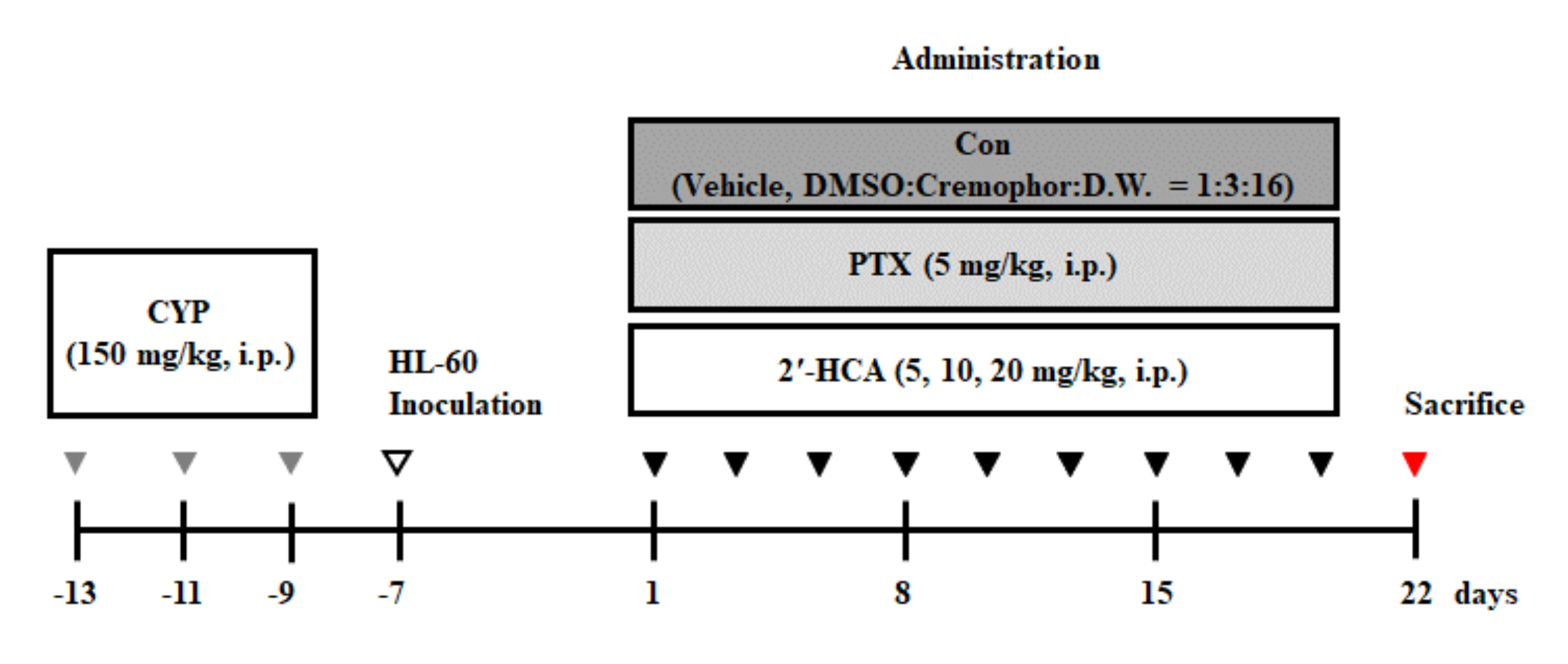
2.15. Animals
2.16. Xenograft Animal Model
2.17. Immunohistochemistry (IHC)
2.18. Statistical Analysis
3. Results
3.1. 2′-HCA Induces Apoptosis in HL-60 Cells
3.2. 2′-HCA-Induced Apoptosis Is Involved in Mitochondrial Dysfunction and Caspase Activations in HL-60 Cells
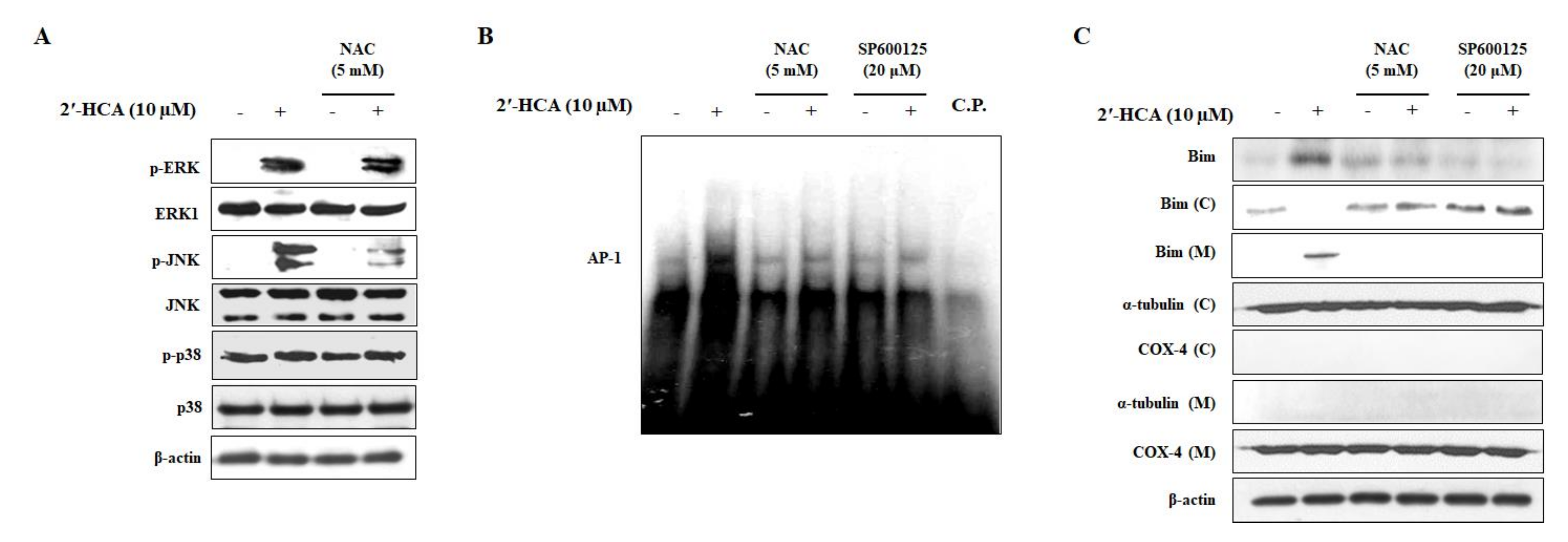
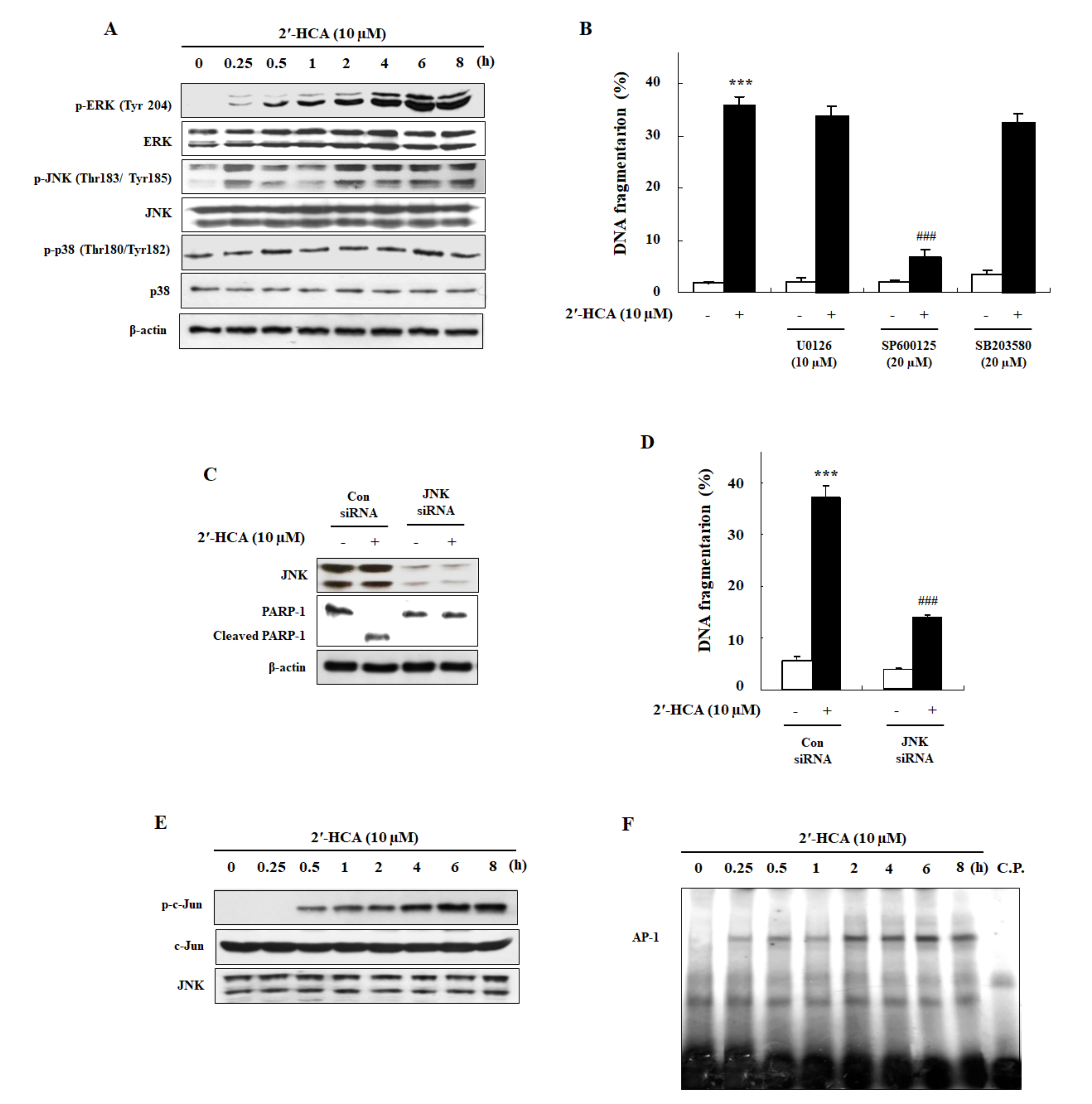
3.3. 2′-HCA-Induced Apoptosis Is Regulated by JNK Activation in HL-60 Cells
3.4. Oxidative Stress Is Required for 2′-HCA-Induced Apoptosis in HL-60 Cells
3.5. Reduced Oxidative Stress Attenuates the 2′-HCA-Induced JNK Pathway and Mitochondrial Translocation of Bim in HL-60 Cells
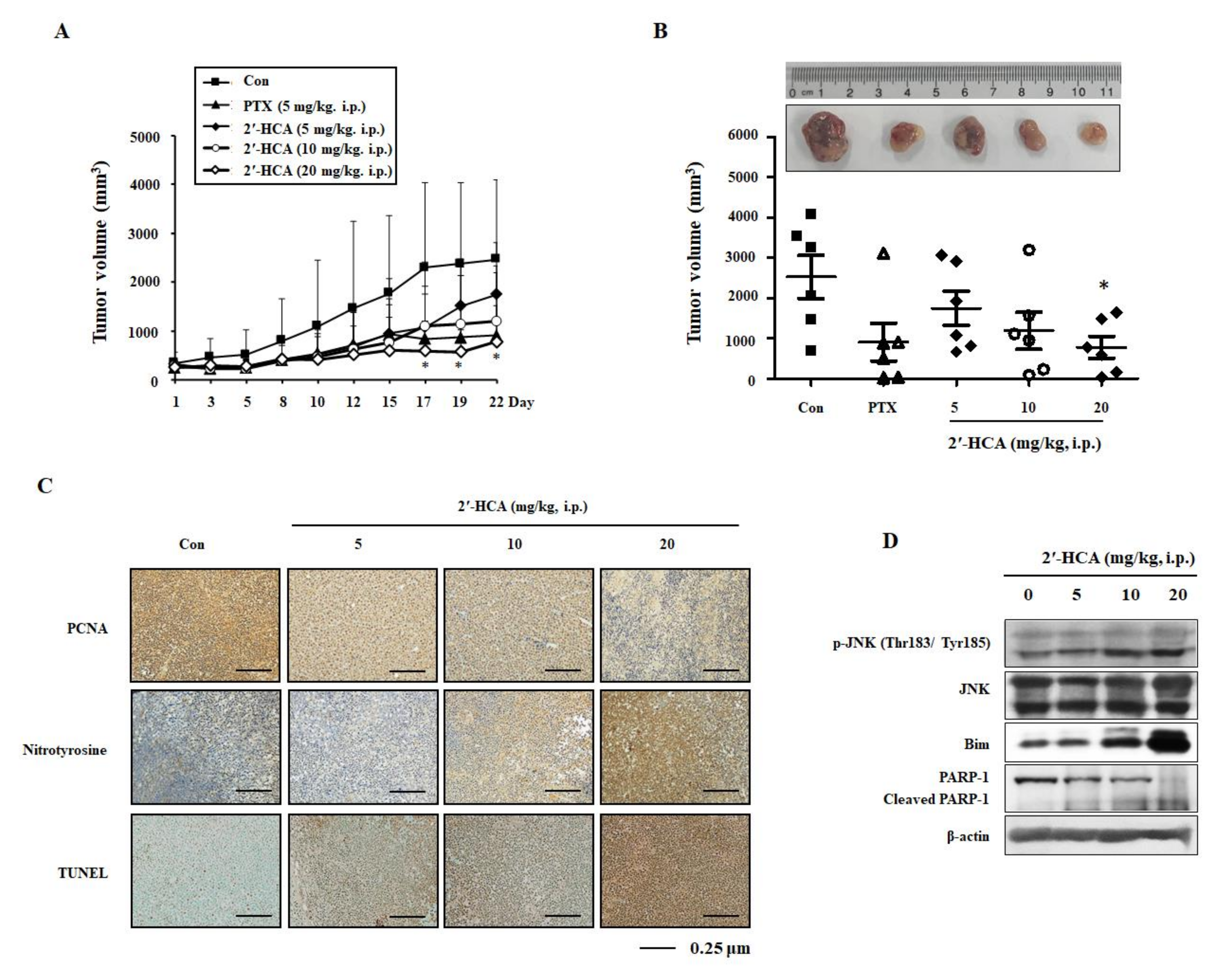
3.6. 2′-HCA Suppresses Tumor Growth in a HL-60 Xenograft Mouse Model
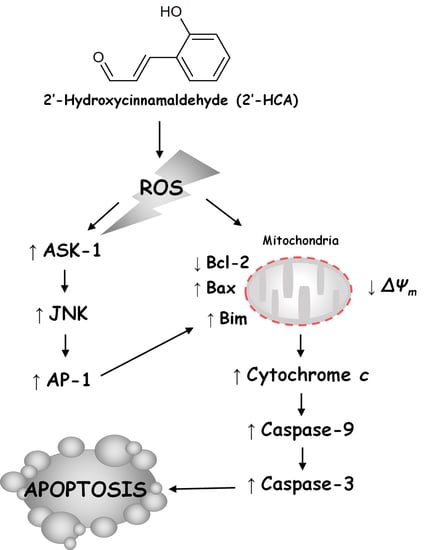
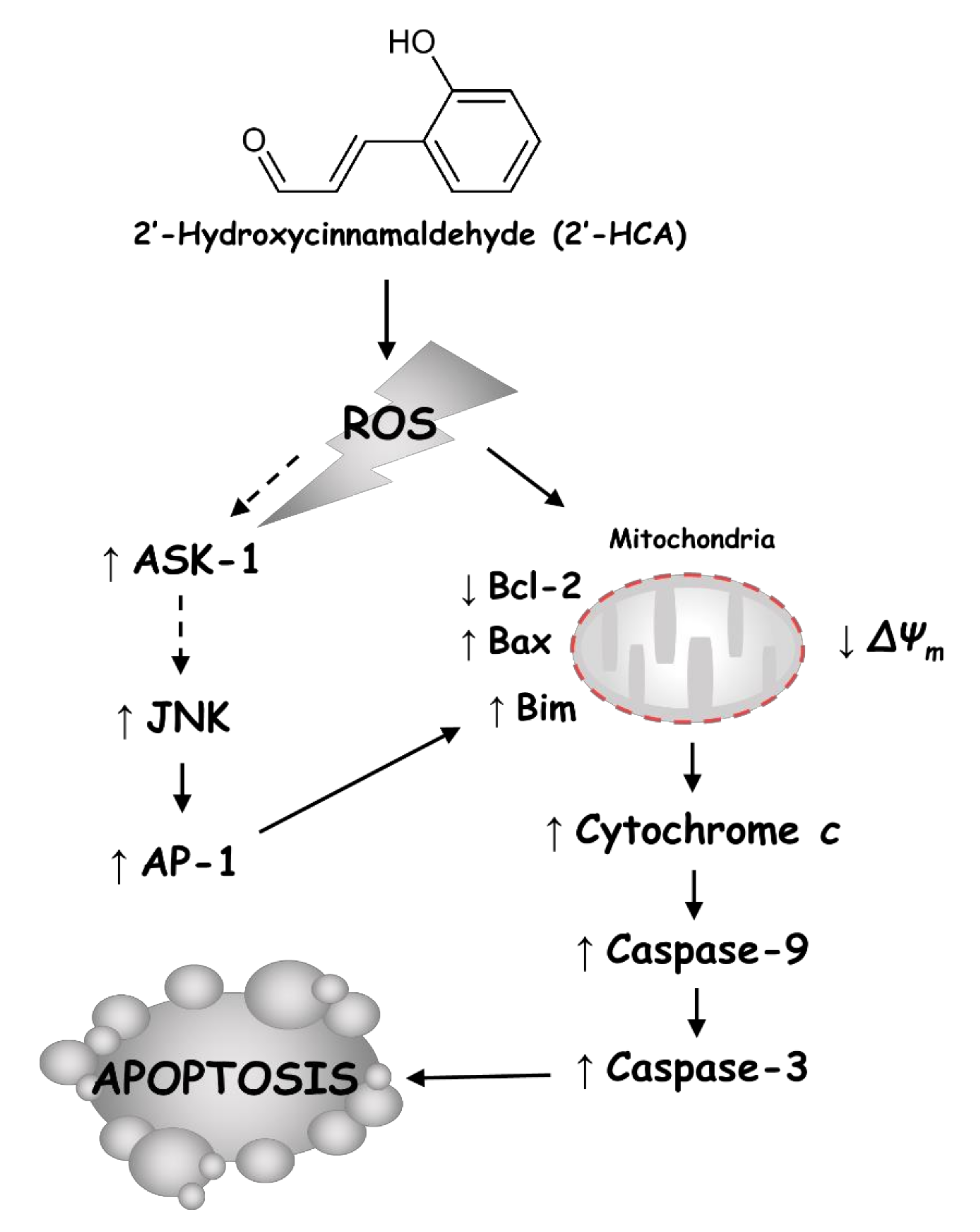
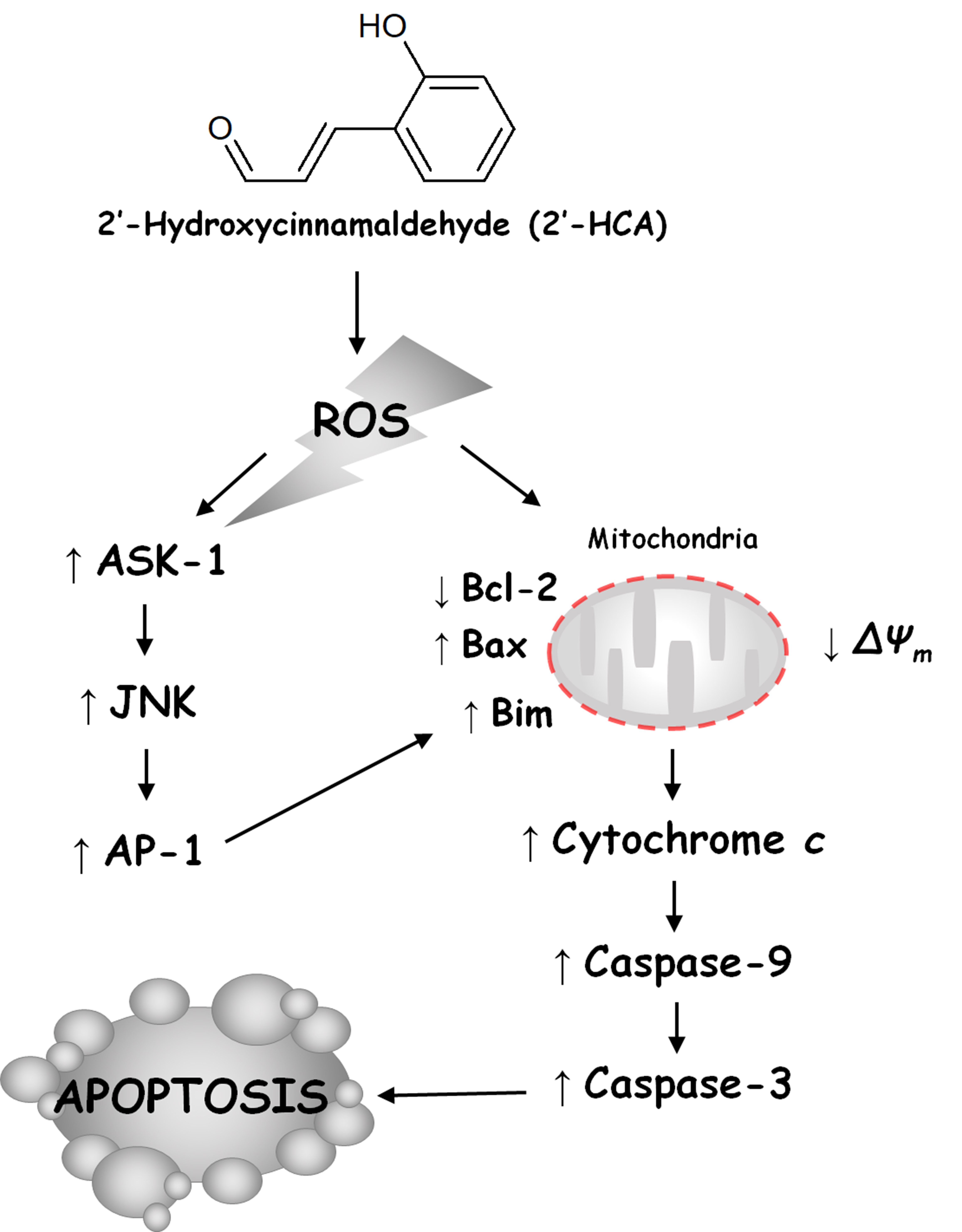
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Pérez, P.S.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Salah, H.T.; Muhsen, I.N.; Salama, M.E.; Owaidah, T.; Hashmi, S.K. Machine learning applications in the diagnosis of leukemia: Current trends and future directions. Int. J. Lab. Hematol. 2019, 41, 717–725. [Google Scholar] [CrossRef]
- Huang, Q.; Wang, L.; Ran, Q.; Wang, J.; Wang, C.; He, H.; Li, L.; Qi, H. Notopterol-induced apoptosis and differentiation in human acute myeloid leukemia HL-60 cells. Drug Des. Dev. Ther. 2019, 13, 1927–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Lim, G.Y.; Hwang, J.; Ryoo, Z.Y.; Huh, T.L.; Lee, S. Induction of apoptosis by obovatol as a novel therapeutic strategy for acute myeloid leukemia. Int. J. Mol. Med. 2014, 34, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Doucette, K.; Norsworthy, K. Recent drug approvals for acute myeloid leukemia. J. Hematol. Oncol. 2019, 12, 100. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, F.; Lv, T.; Chen, Y.; Wen, L.; Zhang, J.; Jiang, X. Apoptosis of HL-60 human leukemia cells induced by Asiatic acid through modulation of B-cell lymphoma 2 family proteins and the mitogen-activated protein kinase signaling pathway. Mol. Med. Rep. 2012, 12, 1429–1434. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.G.; Jin, Y.H.; Yoon, J.H.; Kim, S.A. The anticancer mechanism of 2′-hydroxycinnamaldehyde in human head and neck cancer cells. Int. J. Oncol. 2015, 47, 1793–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Hong, D.; Han, S.; Park, S.; Kim, H.; Kwon, B.-M.; Kim, H. Inhibition of Human Tumor Growth by 2′-Hydroxy- and 2′-Benzoyl-oxycinnamaldehydes. Planta Med. 1999, 65, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Lee, M.Y.; Shin, K.D.; Jeon, S.B.; Kim, J.M.; Son, K.H.; Kim, H.C.; Kim, H.M.; Kwon, B.M. 2′-Benzoyloxycinnamaldehyde Induces Apoptosis in Human Carcinoma via Reactive Oxygen Species. J. Biol. Chem. 2004, 279, 6911–6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.Y.; Lee, M.R.; Wang, A.G.; Lee, J.H.; Kim, H.C.; Kim, H.M.; Kim, J.M.; Kwon, B.M.; Yu, D.Y. Delayed occurrence of H-ras12V-induced hepatocellular carcinoma with long-term treatment with cinnamaldehydes. Eur. J. Pharmacol. 2006, 530, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.A.; Kang, H.S.; Lee, H.J.; Chang, H.; Yun, J.; Lee, C.W.; Kim, N.H.; Kim, H.S.; Yook, J.I.; Hong, S.H.; et al. 2-Hydroxycinnamaldehyde inhibits the epithelial-mesenchymal transition in breast cancer cells. Breast Cancer Res. Treat. 2013, 137, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.J.; Kim, Y.H.; Jin, Y.; Chi, S.W.; Moon, J.H.; Han, D.C.; Kwon, B.M. 2′-hydroxycinnamaldehyde inhibits cancer cell proliferation and tumor growth by targeting the pyruvate kinase M2. Cancer Lett. 2018, 434, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.J.; Kim, Y.H.; Lee, Y.J.; Choi, J.; Kim, C.H.; Han, D.C.; Kwon, B.M. 2′-Hydroxycinnamaldehyde inhibits proliferation and induces apoptosis via signal transducer and activator of transcription 3 inactivation and reactive oxygen species generation. Cancer Sci. 2019, 110, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.M.; Cho, Y.K.; Lee, S.H.; Nam, J.Y.; Bok, S.H.; Chun, S.K.; Kim, J.A.; Lee, I.R. 2′-Hydroxycinnamaldehyde from Stem Bark ofCinnamomum cassia. Planta Med. 1996, 62, 183–184. [Google Scholar] [CrossRef]
- Plumb, J.A.; Milroy, R.; Kaye, S.B. Effects of the pH dependence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide-formazan absorption on chemosensitivity determined by a novel tetrazolium-based assay. Cancer Res. 1989, 49, 4435–4440. [Google Scholar]
- Watabe, M.; Kawazoe, N.; Masuda, Y.; Nakajo, S.; Nakaya, K. Bcl-2 protein inhibits bufalin-induced apoptosis through inhi-bition of mitogen-activated protein kinase activation in human leukemia U937 cells. Cancer Res 1997, 57, 3097–3100. [Google Scholar] [PubMed]
- Lee, K.W.; Jung, H.J.; Park, H.J.; Kim, D.G.; Lee, J.Y.; Lee, K.T. Beta-D-xylopyranosyl-(1-->3)-beta-D-glucuronopyranosyl echinocystic acid isolated from the roots of Codonopsis lanceolata induces caspase-dependent apoptosis in human acute promyelocytic leukemia HL-60 cells. Biol. Pharm. Bull. 2005, 28, 854–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavelescu, L.A. On reactive oxygen species measurement in living systems. J. Med. Life 2015, 8, 38–42. [Google Scholar]
- Eady, J.J.; Orta, T.; Dennis, M.F.; Stratford, M.R.; Peacock, J.H. Glutathione determination by the Tietze enzymatic recycling assay and its relationship to cellular radiation response. Br. J. Cancer 1995, 72, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Ha, J.; Park, J.H.; Lee, J.Y.; Lee, Y.S.; Park, H.J.; Choi, J.W.; Masuda, Y.; Nakaya, K.; Lee, K.T. Costunolide triggers apoptosis in human leukemia U937 cells by depleting intracellular thiols. Jpn. J. Cancer Res. 2002, 93, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Park, S.J.; Ryu, S.; Kang, H.B.; Kim, T.W.; Choi, J.H.; Lee, J.Y.; Cho, Y.W.; Lee, K.T. Potent anti-inflammatory effect of a novel furan-2,5-dione derivative, BPD, mediated by dual suppression of COX-2 activity and LPS-induced inflammatory gene expression via NF-κB inactivation. Br. J. Pharmacol. 2012, 165, 1926–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.Y.; Chung, K.S.; Shin, J.S.; Lee, J.H.; Gil, H.S.; Lee, H.H.; Choi, E.; Choi, J.H.; Hassan, A.H.E.; Lee, Y.S.; et al. The Anti-Proliferative Activity of the Hybrid TMS-TMF-4f Compound Against Human Cervical Cancer Involves Apoptosis Mediated by STAT3 Inactivation. Cancers 2019, 11, 1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Gan, D.; Huang, Q.; Luo, X.; Lin, D.; Hu, J. Emodin and Its Combination with Cytarabine Induce Apoptosis in Resistant Acute Myeloid Leukemia Cells in Vitro and in Vivo. Cell. Physiol. Biochem. 2018, 48, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive Oxygen and Nitrogen Species–Induced Protein Modifications: Implication in Carcinogenesis and Anticancer Therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziech, D.; Franco, R.; Georgakilas, A.G.; Georgakila, S.; Malamou-Mitsi, V.; Schoneveld, O.; Pappa, A.; Panayiotidis, M.I. The role of reactive oxygen species and oxidative stress in environmental carcinogenesis and biomarker development. Chem. Interact. 2010, 188, 334–339. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, D.; Lee, K.W.; Yoon, D.; Surh, Y. Jaceosidin Induces Apoptosis in ras-Transformed Human Breast Epithelial Cells through Generation of Reactive Oxygen Species. Ann. N. Y. Acad. Sci. 2007, 1095, 483–495. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, Y.J.; Lee, S.A.; Myung, S.C.; Kim, W. Combined Effect of Hsp90 Inhibitor Geldanamycin and Parthenolide via Reactive Oxygen Species-Mediated Apoptotic Process on Epithelial Ovarian Cancer Cells. Basic Clin. Pharmacol. Toxicol. 2012, 111, 173–181. [Google Scholar] [CrossRef]
- Choi, J.H.; Lee, K.T. Costunolide-Induced Apoptosis in Human Leukemia Cells: Involvement of c-Jun N-Terminal Kinase Activation. Biol. Pharm. Bull. 2009, 32, 1803–1808. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.I.; Kim, J.H.; Lee, K.T.; Choi, J.H. Costunolide induces apoptosis in platinum-resistant human ovarian cancer cells by generating reactive oxygen species. Gynecol. Oncol. 2011, 123, 588–596. [Google Scholar] [CrossRef]
- Ka, H.; Park, H.J.; Jung, H.J.; Choi, J.W.; Cho, K.S.; Ha, J.; Lee, K.T. Cinnamaldehyde induces apoptosis by ROS-mediated mitochondrial permeability transition in human promyelocytic leukemia HL-60 cells. Cancer Lett. 2003, 196, 143–152. [Google Scholar] [CrossRef]
- Kim, J.R.; Son, J.E.; Jeong, H.; Kim, D.J.; Seo, S.G.; Lee, E.; Lim, T.G.; Kimbung, Y.R.; Chen, H.; Bode, A.M.; et al. A Novel Cinnamon-Related Natural Product with Pim-1 Inhibitory Activity Inhibits Leukemia and Skin Cancer. Cancer Res. 2015, 75, 2716–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Lee, S.Y.; Son, D.J.; Lee, H.; Yoo, H.S.; Song, S.; Oh, K.W.; Han, D.C.; Kwon, B.M.; Hong, J.T. Inhibitory effect of 2′-hydroxycinnamaldehyde on nitric oxide production through inhibition of NF-κB activation in RAW 264.7 cells. Biochem. Pharmacol. 2005, 69, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH Oxidase-Mediated Redox Signaling: Roles in Cellular Stress Response, Stress Tolerance, and Tissue Repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 897–906. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susnow, N.; Zeng, L.; Margineantu, D.; Hockenbery, D.M. Bcl-2 family proteins as regulators of oxidative stress. Semin. Cancer Biol. 2009, 19, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Xiang, G.H.; Tang, T.; Tang, Y.; Zhao, L.Y.; Liu, D.; Zhang, Y.R.; Tang, J.T.; Zhou, S.; Wu, D.H. Capsaicin and dihydrocapsaicin induce apoptosis in human glioma cells via ROS and Ca2+-mediated mitochondrial pathway. Mol. Med. Rep. 2016, 14, 4198–4208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.; Chen, Y.; Ding, Z.; Luo, G.; Liu, J.; Shen, J. Novel Insights into the Synergistic Interaction of a Thioredoxin Reductase Inhibitor and TRAIL: The Activation of the ASK1-ERK-Sp1 Pathway. PLoS ONE 2013, 8, e63966. [Google Scholar] [CrossRef] [Green Version]
- Nagai, H.; Noguchi, T.; Takeda, K.; Ichijo, H. Pathophysiological Roles of ASK1-MAP Kinase Signaling Pathways. J. Biochem. Mol. Biol. 2007, 40, 1–6. [Google Scholar] [CrossRef]
- Gu, J.J.; Wang, Z.; Reeves, R.; Magnuson, N.S. PIM1 phosphorylates and negatively regulates ASK1-mediated apoptosis. Oncogene 2009, 28, 4261–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Peuckert, C.; Wölfl, S. Essential role of mitochondrial Stat3 in p38MAPK mediated apoptosis under oxidative stress. Sci. Rep. 2017, 7, 15388. [Google Scholar] [CrossRef]
- Shen, H.M.; Liu, Z.G. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free. Radic. Biol. Med. 2006, 40, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Abe, J.; Haendeler, J.; Huang, Q.; Berk, B.C. Src and Cas Mediate JNK Activation but Not ERK1/2 and p38 Kinases by Reactive Oxygen Species. J. Biol. Chem. 2000, 275, 11706–11712. [Google Scholar] [CrossRef] [Green Version]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK Activation in Cisplatin-induced Apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef] [Green Version]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Kasibhatla, S.; Brunner, T.; Genestier, L.; Echeverri, F.; Mahboubi, A.; Green, D.R. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol. Cell 1998, 1, 543–551. [Google Scholar] [CrossRef]
- Ameyar, M.; Wisniewska, M.; Weitzman, J. A role for AP-1 in apoptosis: The case for and against. Biochimie 2003, 85, 747–752. [Google Scholar] [CrossRef]
- Whitfield, J.; Neame, S.J.; Paquet, L.; Bernard, O.; Ham, J. Dominant-Negative c-Jun Promotes Neuronal Survival by Reducing BIM Expression and Inhibiting Mitochondrial Cytochrome c Release. Neuron 2001, 29, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Yao, C.F.; Huang, S.M.; Ko, S.; Tan, Y.H.; Lee-Chen, G.J.; Wang, Y.C. Novel 2-step synthetic indole compound 1,1,3-tri(3-indolyl)cyclohexane inhibits cancer cell growth in lung cancer cells and xenograft models. Cancer 2008, 113, 815–825. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Origin | IC50 (μM) (a) | |
|---|---|---|---|
| 2′-HCA | Cisplatin | ||
| HL-60 | Human promyelocytic leukemia | 4.17 ± 0.62 | 17.7 ± 2.54 |
| Molt-4 | Human T lymphoblastic leukemia | 9.14 ± 0.88 | 9.34 ± 1.12 |
| U937 | Human histiocytic lymphoma | 23.25 ± 1.98 | 22.0 ± 2.18 |
| K562 | Human erythroleukemia | 23.15 ± 2.34 | >100 |
| HepG2 | Human hepatoblastoma | 17.08 ± 1.76 | 54.6 ± 4.88 |
| SNU-C5 | Human colorectal adenocarcinoma | 22.77 ± 2.92 | 27.4 ± 2.76 |
| A549 | Human lung adenocarcinoma | 33.14 ± 3.76 | 47.62 ± 3.87 |
| KB | Human mouth epidermal carcinoma | 53.04 ± 4.91 | 56.43 ± 4.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, K.-S.; Yoo, C.-B.; Lee, J.-H.; Lee, H.-H.; Park, S.-E.; Han, H.-S.; Lee, S.-Y.; Kwon, B.-M.; Choi, J.-H.; Lee, K.-T. Regulation of ROS-Dependent JNK Pathway by 2’-Hydroxycinnamaldehyde Inducing Apoptosis in Human Promyelocytic HL-60 Leukemia Cells. Pharmaceutics 2021, 13, 1794. https://doi.org/10.3390/pharmaceutics13111794
Chung K-S, Yoo C-B, Lee J-H, Lee H-H, Park S-E, Han H-S, Lee S-Y, Kwon B-M, Choi J-H, Lee K-T. Regulation of ROS-Dependent JNK Pathway by 2’-Hydroxycinnamaldehyde Inducing Apoptosis in Human Promyelocytic HL-60 Leukemia Cells. Pharmaceutics. 2021; 13(11):1794. https://doi.org/10.3390/pharmaceutics13111794
Chicago/Turabian StyleChung, Kyung-Sook, Chae-Bin Yoo, Jeong-Hun Lee, Hwi-Ho Lee, Sang-Eun Park, Hee-Soo Han, Su-Yeon Lee, Byoung-Mok Kwon, Jung-Hye Choi, and Kyung-Tae Lee. 2021. "Regulation of ROS-Dependent JNK Pathway by 2’-Hydroxycinnamaldehyde Inducing Apoptosis in Human Promyelocytic HL-60 Leukemia Cells" Pharmaceutics 13, no. 11: 1794. https://doi.org/10.3390/pharmaceutics13111794