
Folate Functionalized Lipid Nanoparticles for Targeted Therapy of Methicillin-Resistant Staphylococcus aureus

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
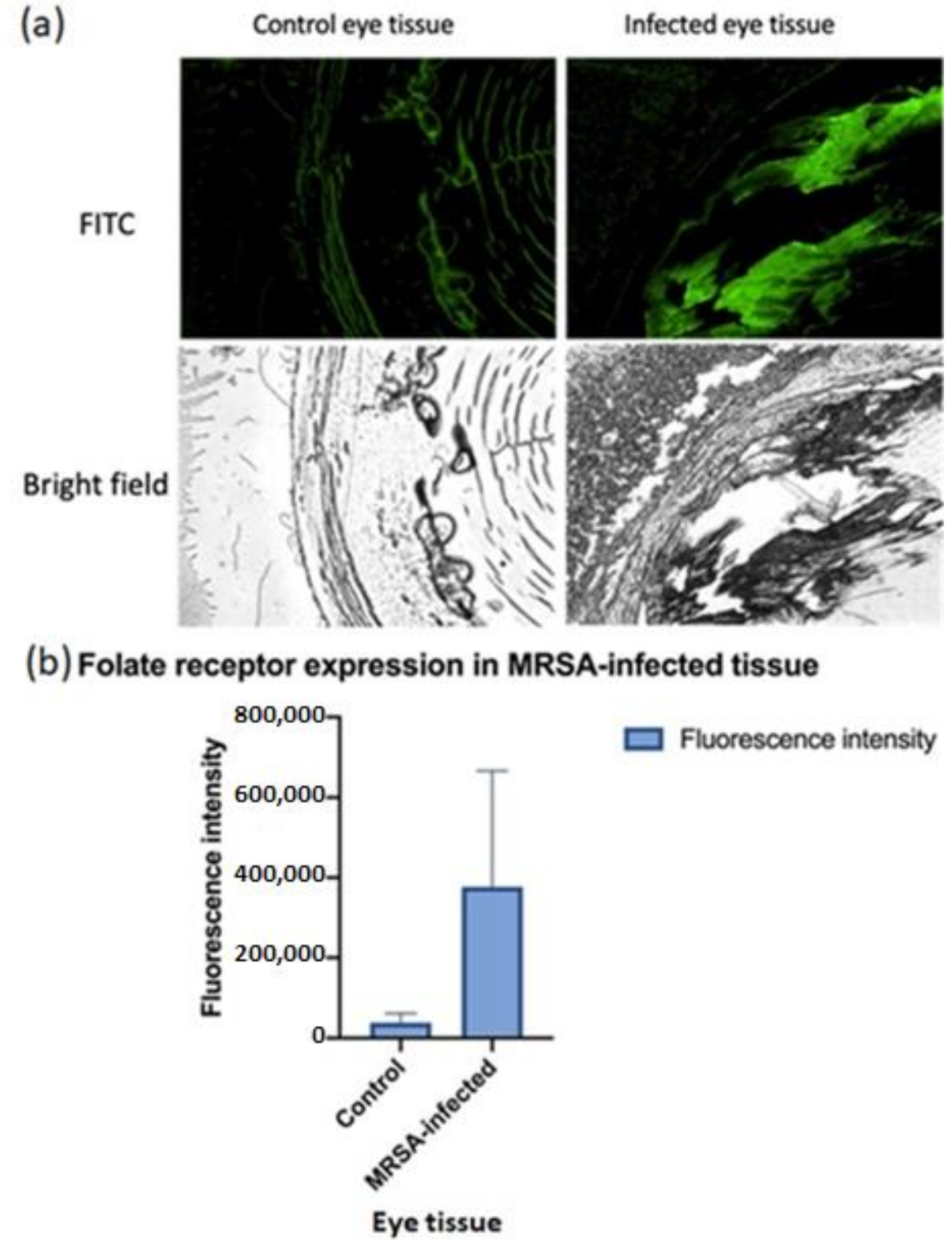
2.2. Immunohistochemistry for Folate Receptor Expression
2.3. Formulation Development of LVAN
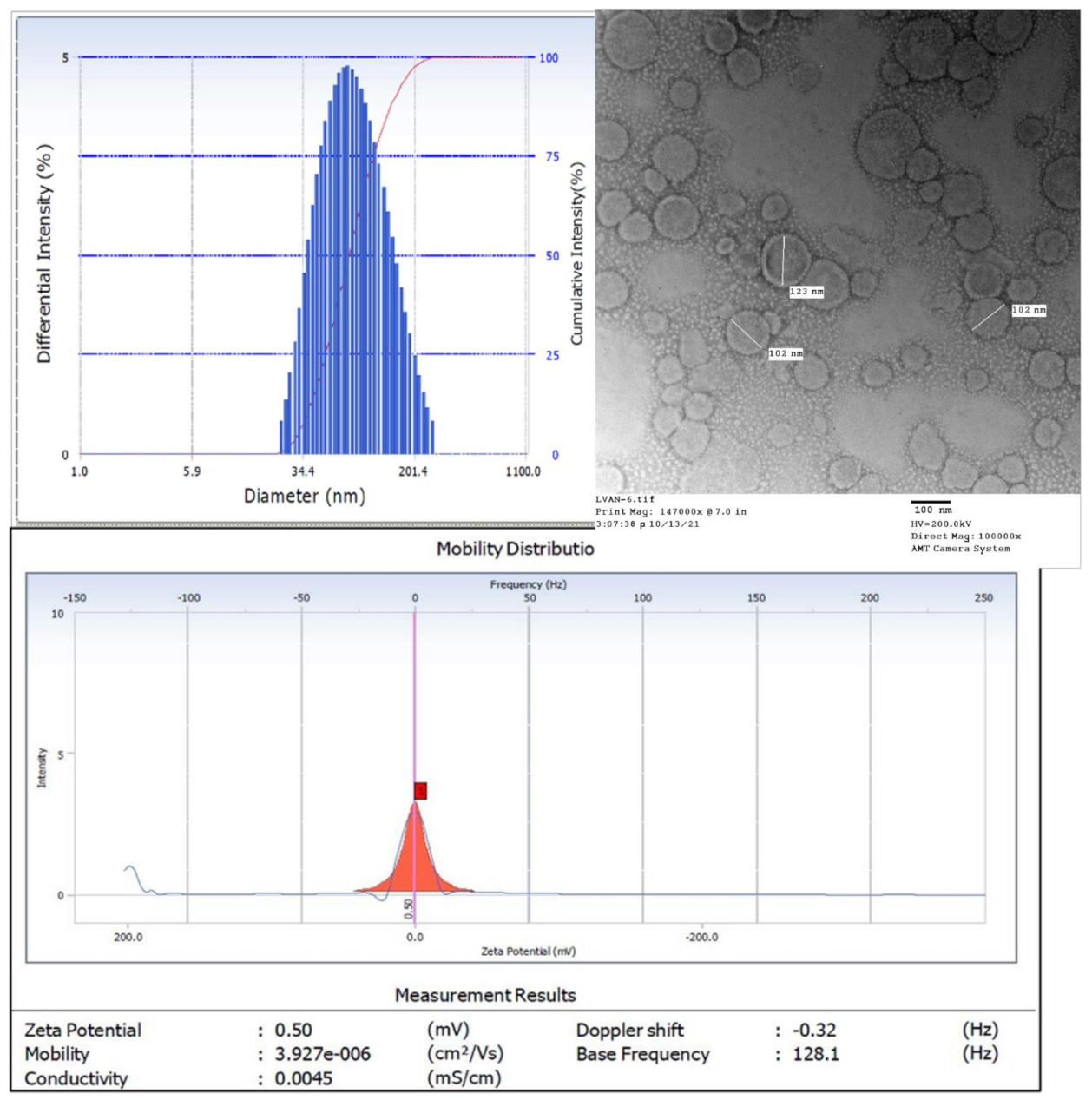
2.4. Physicochemical Characterization
2.4.1. Size, Polydispersity Index, and Zeta Potential
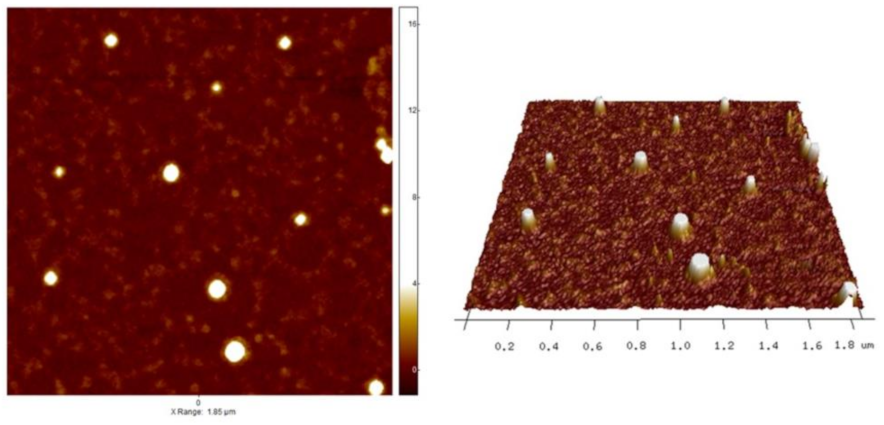
2.4.2. Atomic Force Microscopy
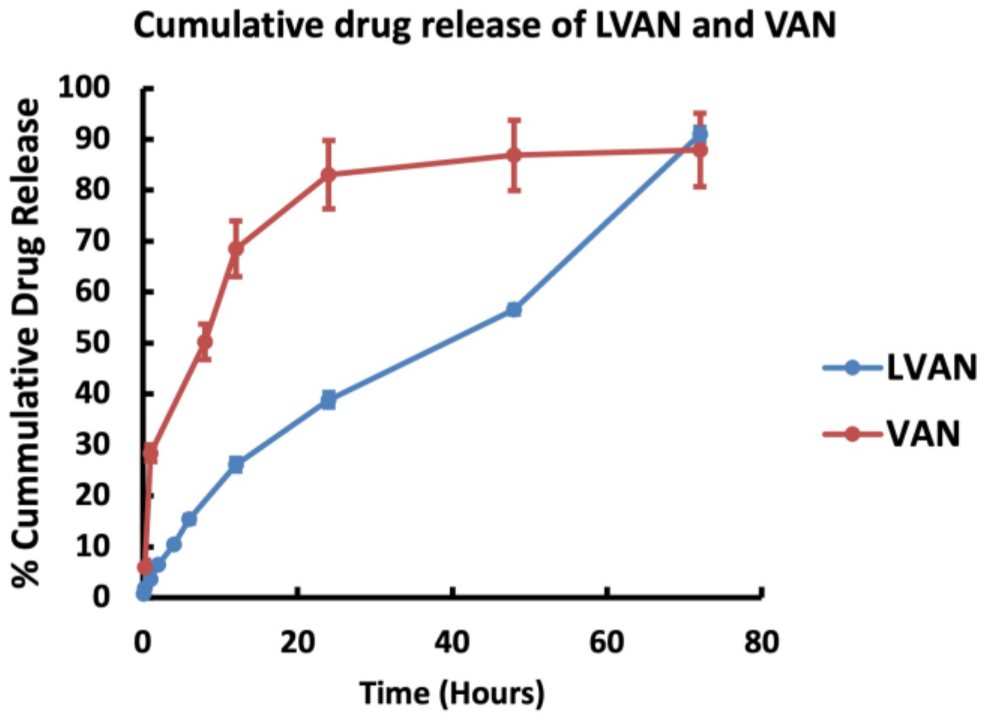
2.4.3. In Vitro Drug Release Study
2.5. Susceptibility Testing
2.6. In Vitro Disk Diffusion Assay
2.7. Biofilm Inhibition of LVAN vs. VAN
2.8. In Vivo Testing on Infected Mouse Thigh Model
2.9. In Vivo Tissue Uptake of LVAN vs. VAN
2.10. In Vivo Antibacterial Efficacy Study
3. Results and Discussion
3.1. Folate Receptor Expression
3.2. Formulation Development and Physicochemical Characterization
3.2.1. Size and Morphology
3.2.2. Atomic Force Microscopy
3.2.3. In Vitro Drug Release
3.3. Susceptibility Testing
3.4. In Vitro Disk Diffusion Assay
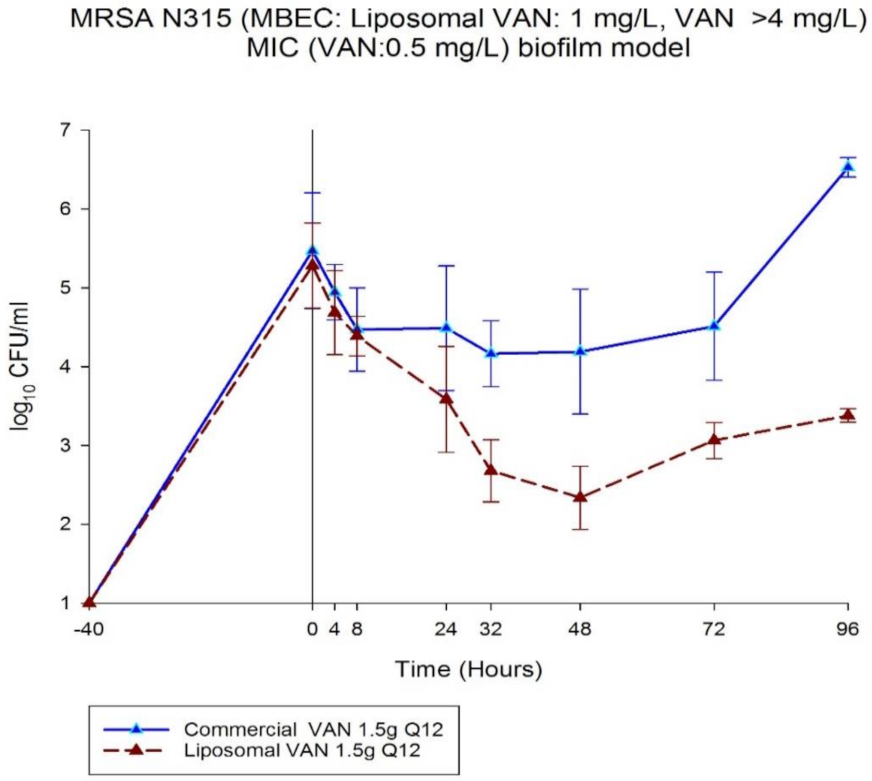
3.5. Biofilm Inhibition of LVAN vs. VAN
3.6. In Vivo Testing on Infected Mouse Thigh Model
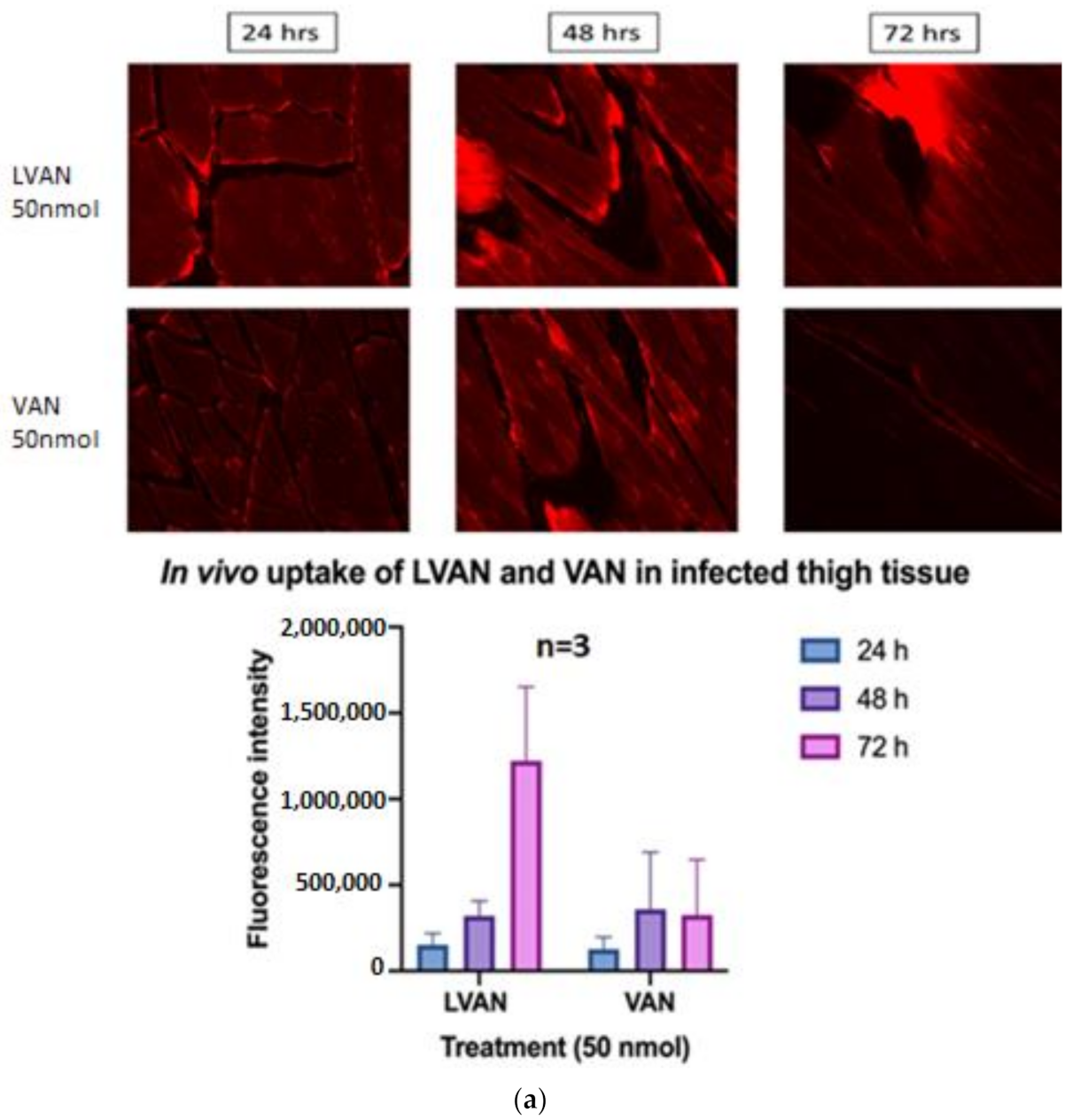
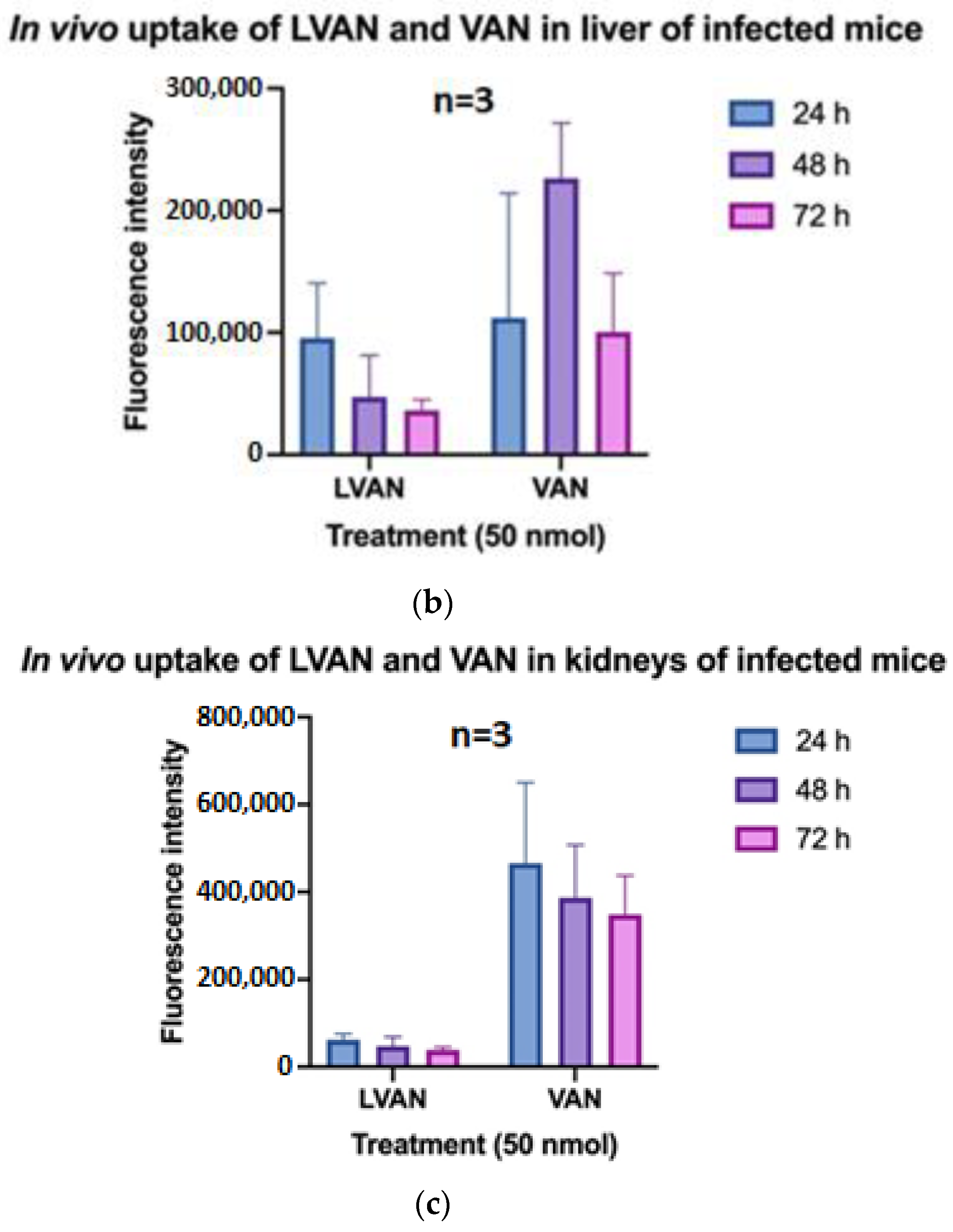
3.6.1. In Vivo Tissue Uptake of LVAN vs. VAN
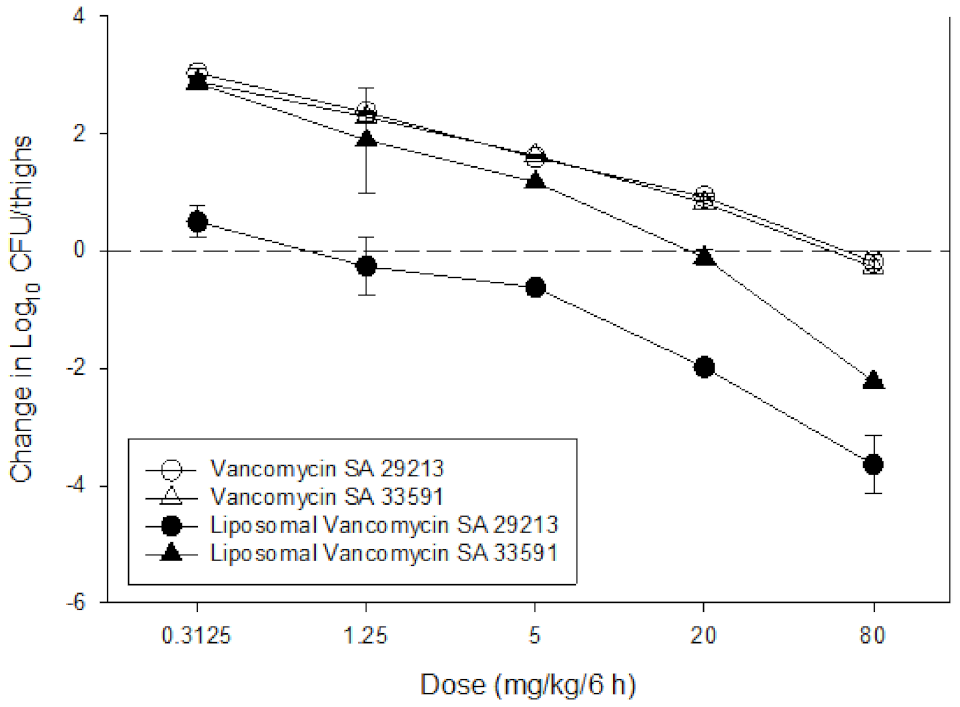
3.6.2. In Vivo Antibacterial Efficacy Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Vanamala, K.; Tatiparti, K.; Bhise, K.; Sau, S.; Scheetz, M.H.; Rybak, M.J.; Andes, D.; Iyer, A.K. Novel approaches for the treatment of methicillin-resistant Staphylococcus aureus: Using nanoparticles to overcome multidrug resistance. Drug Discov. Today 2021, 26, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; David, M.Z.; Boyle-Vavra, S.; Sieth, J.; Daum, R.S. High Staphylococcus aureus colonization prevalence among patients with skin and soft tissue infections and controls in an urban emergency department. J. Clin. Microbiol. 2015, 53, 810–815. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.T. Control of MSSA and MRSA in the United States: Protocols, policies, risk adjustment and excuses. Antimicrob. Resist. Infect. Control 2019, 8, 103. [Google Scholar] [CrossRef]
- Davis, K.A.; Stewart, J.J.; Crouch, H.K.; Florez, C.E.; Hospenthal, D.R. Methicillin-Resistant Staphylococcus aureus (MRSA) Nares Colonization at Hospital Admission and Its Effect on Subsequent MRSA Infection. Clin. Infect. Dis. 2004, 39, 776–782. [Google Scholar] [CrossRef]
- Davis, S.L.; Rybak, M.J.; Amjad, M.; Kaatz, G.W.; McKinnon, P.S. Characteristics of Patients With Healthcare-Associated Infection Due to SCC mec Type IV Methicillin-Resistant Staphylococcus aureus. Infect. Control Hosp. Epidemiol. 2006, 27, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: Executive summary. Clin. Infect. Dis. 2011, 52, 285–292. [Google Scholar] [CrossRef]
- Kollef, M.H. Limitations of vancomycin in the management of resistant staphylococcal infections. Clin. Infect. Dis. 2007, 45, 191–195. [Google Scholar] [CrossRef]
- Hassoun, A.; Linden, P.K.; Friedman, B. Incidence, prevalence, and management of MRSA bacteremia across patient populations-a review of recent developments in MRSA management and treatment. Crit. Care 2017, 21, 211. [Google Scholar] [CrossRef] [Green Version]
- Meaney, C.J.; Hynicka, L.M.; Tsoukleris, M.G. Vancomycin-associated nephrotoxicity in adult medicine patients: Incidence, outcomes, and risk factors. Pharmacotherapy 2014, 34, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Barber, K.E.; Werth, B.J.; McRoberts, J.P.; Rybaka, M.J. A novel approach utilizing biofilm time-kill curves to assess the bactericidal activity of ceftaroline combinations against biofilm- producing methicillin-resistant staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 2989–2992. [Google Scholar] [CrossRef] [Green Version]
- Khardori, N.; Yassien, M. Biofilms in device-related infections. J. Ind. Microbiol. 1995, 15, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Cully, M. Antibiotics alter the gut microbiome and host health. Nat. Milest. 2019, 1423, S19. [Google Scholar]
- Borsa, B.A.; Sudagidan, M.; Aldag, M.E.; Baris, I.I.; Acar, E.E.; Acuner, C.; Kavruk, M.; Ozalp, V.C. Antibiotic administration in targeted nanoparticles protects the faecal microbiota of mice. RSC Med. Chem. 2021, 12, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Cross, R. Without these lipid shells, there would be no mRNA vaccines for COVID-19. Chem. Eng. News 2021, 16–19. [Google Scholar] [CrossRef]
- Bhise, K.; Kashaw, S.K.; Sau, S.; Iyer, A.K. Nanostructured Lipid Carriers Employing Polyphenols as Promising Anticancer Agents: Quality by Design (QbD) approach. Int. J. Pharm. 2017, 526, 506–515. [Google Scholar] [CrossRef]
- Bhise, K.; Sau, S.; Alsaab, H.; Kashaw, S.K.; Tekade, R.K.; Iyer, A.K. Nanomedicine for cancer diagnosis and therapy: Advancement, success and structure-activity relationship. Ther. Deliv. 2017, 8, 1003–1018. [Google Scholar] [CrossRef]
- Sau, S.; Agarwalla, P.; Mukherjee, S.; Bag, I.; Sreedhar, B.; Pal-Bhadra, M.; Patra, C.R.; Banerjee, R. Cancer cell-selective promoter recognition accompanies antitumor effect by glucocorticoid receptor-targeted gold nanoparticle. Nanoscale 2014, 6, 6745–6754. [Google Scholar] [CrossRef]
- Gawde, K.A.; Kesharwani, P.; Sau, S.; Sarkar, F.H.; Padhye, S.; Kashaw, S.K.; Iyer, A.K. Synthesis and characterization of folate decorated albumin bio-conjugate nanoparticles loaded with a synthetic curcumin difluorinated analogue. J. Colloid Interface Sci. 2017, 496, 290–299. [Google Scholar] [CrossRef]
- Sahu, P.; Kashaw, S.K.; Jain, S.; Sau, S.; Iyer, A.K. Assessment of penetration potential of pH responsive double walled biodegradable nanogels coated with eucalyptus oil for the controlled delivery of 5-fluorouracil: In vitro and ex vivo studies. J. Control. Release 2017, 253, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sau, S.; Alsaab, H.O.; Iyer, A.K. CD44 directed nanomicellar payload delivery platform for selective anticancer effect and tumor specific imaging of triple negative breast cancer. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1441–1454. [Google Scholar] [CrossRef]
- Bhise, K.; Sau, S.; Kebriaei, R.; Rice, S.A.; Stamper, K.C.; Alsaab, H.O.; Rybak, M.J.; Iyer, A.K. Combination of Vancomycin and Cefazolin Lipid Nanoparticles for Overcoming Antibiotic Resistance of MRSA. Materials 2018, 11, 1245. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Putt, K.S.; Visscher, D.W.; Murphy, L.; Cohen, C.; Singhal, S.; Sandusky, G.; Feng, Y.; Dimitrov, D.S.; Low, P.S. Assessment of folate receptor-β expression in human neoplastic tissues. Oncotarget 2015, 6, 14700–14709. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Zaynagetdinov, R.; Yull, F.E.; Polosukhin, V.V.; Gleaves, L.A.; Tanjore, H.; Young, L.R.; Peterson, T.E.; Manning, H.C.; Prince, L.S.; et al. Molecular imaging of folate receptor β-positive macrophages during acute lung inflammation. Am. J. Respir. Cell Mol. Biol. 2015, 53, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Lepak, A.J.; Andes, D.R. Animal models in the pharmacokinetic/pharmacodynamic evaluation of antimicrobial agents. Bioorg. Med. Chem. 2016, 24, 6390–6400. [Google Scholar] [CrossRef]
- Andes, D.R.; Lepak, A.J. In vivo infection models in the pre-clinical pharmacokinetic/pharmacodynamic evaluation of antimicrobial agents. Curr. Opin. Pharmacol. 2017, 36, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Lepak, A.J.; Zhao, M.; Marchillo, K.; VanHecker, J.; Andes, D.R. In Vivo Pharmacodynamics of Omadacycline against Staphylococcus aureus in the Neutropenic Murine Thigh Infection Model. Antimicrob. Agents Chemother. 2019, 63, 22–25. [Google Scholar] [CrossRef] [Green Version]
- Puig-Kröger, A.; Sierra-Filardi, E.; Domínguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gómez-Aguado, F.; Ratnam, M.; Sánchez-Mateos, P.; Corbí, A.L. Folate receptor β is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory Macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggl, M.; Pate, V.; Stürmer, T.; Kshirsagar, A.V.; Layton, J.B. The comparative risk of acute kidney injury of vancomycin relative to other common antibiotics. Sci. Rep. 2020, 10, 17282. [Google Scholar] [CrossRef] [PubMed]
- Nolin, T.D. Vancomycin and the risk of AKI: Now clearer than mississippi mud. Clin. J. Am. Soc. Nephrol. 2016, 11, 2101–2103. [Google Scholar] [CrossRef] [PubMed]
- Filippone, E.J.; Kraft, W.K.; Farber, J.L. The Nephrotoxicity of Vancomycin. Clin. Pharmacol. Ther. 2017, 102, 459–469. [Google Scholar] [CrossRef]
- Kim, B.; Pang, H.B.; Kang, J.; Park, J.H.; Ruoslahti, E.; Sailor, M.J. Immunogene therapy with fusogenic nanoparticles modulates macrophage response to Staphylococcus aureus. Nat. Commun. 2018, 9, 1969. [Google Scholar] [CrossRef]
- Wang, L.; Hu, C.; Shao, L. The antimicrobial activity of nanoparticles: Present situation and prospects for the future. Int. J. Nanomed. 2017, 12, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahbubul, I.M.; Shahrul, I.M.; Khaleduzzaman, S.S.; Saidur, R.; Amalina, M.A.; Turgut, A. Experimental investigation on effect of ultrasonication duration on colloidal dispersion and thermophysical properties of alumina-water nanofluid. Int. J. Heat Mass Transf. 2015, 88, 73–81. [Google Scholar] [CrossRef]
- Pumerantz, A.; Muppidi, K.; Agnihotri, S.; Guerra, C.; Venketaraman, V.; Wang, J.; Betageri, G. Preparation of liposomal vancomycin and intracellular killing of meticillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 2011, 37, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Rybak, M.J. The pharmacokinetic and pharmacodynamic properties of vancomycin. Clin. Infect. Dis. 2006, 42 (Suppl. 1), S35–S39. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | MIC Values (mg/L) | |
|---|---|---|
| VAN | LVAN | |
| 494 | 1 | 0.5–1 |
| 29213 | 1 | 0.5–1 |
| N315 | 0.5 | 0.5 |
| Strain | Zone of Inhibition (mm) | |
|---|---|---|
| VAN | LVAN | |
| 494 | 10.86 | 11.66 |
| 29213 | 10.37 | 11 |
| N315 | 12.86 | 12.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanamala, K.; Bhise, K.; Sanchez, H.; Kebriaei, R.; Luong, D.; Sau, S.; Abdelhady, H.; Rybak, M.J.; Andes, D.; Iyer, A.K. Folate Functionalized Lipid Nanoparticles for Targeted Therapy of Methicillin-Resistant Staphylococcus aureus. Pharmaceutics 2021, 13, 1791. https://doi.org/10.3390/pharmaceutics13111791
Vanamala K, Bhise K, Sanchez H, Kebriaei R, Luong D, Sau S, Abdelhady H, Rybak MJ, Andes D, Iyer AK. Folate Functionalized Lipid Nanoparticles for Targeted Therapy of Methicillin-Resistant Staphylococcus aureus. Pharmaceutics. 2021; 13(11):1791. https://doi.org/10.3390/pharmaceutics13111791
Chicago/Turabian StyleVanamala, Kushal, Ketki Bhise, Hiram Sanchez, Razieh Kebriaei, Duy Luong, Samaresh Sau, Hosam Abdelhady, Michael J. Rybak, David Andes, and Arun K. Iyer. 2021. "Folate Functionalized Lipid Nanoparticles for Targeted Therapy of Methicillin-Resistant Staphylococcus aureus" Pharmaceutics 13, no. 11: 1791. https://doi.org/10.3390/pharmaceutics13111791