
Fixed-Dose Combination Formulations in Solid Oral Drug


Abstract
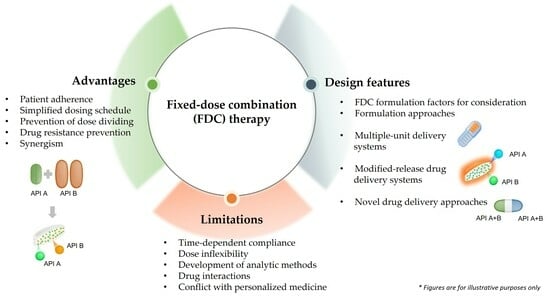
:
1. Introduction
2. Fixed-Dose Combination Formulations in Drug Therapy
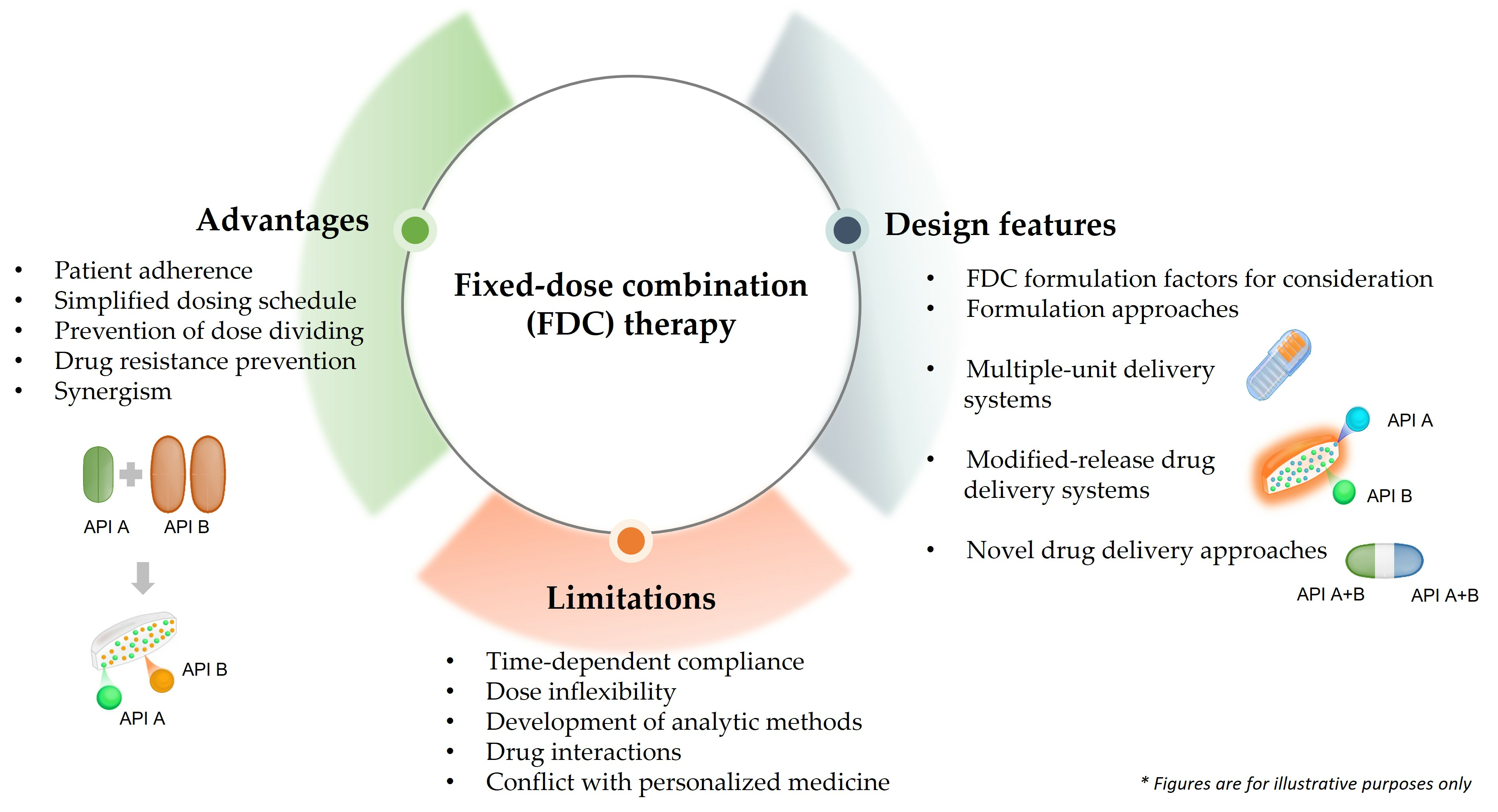
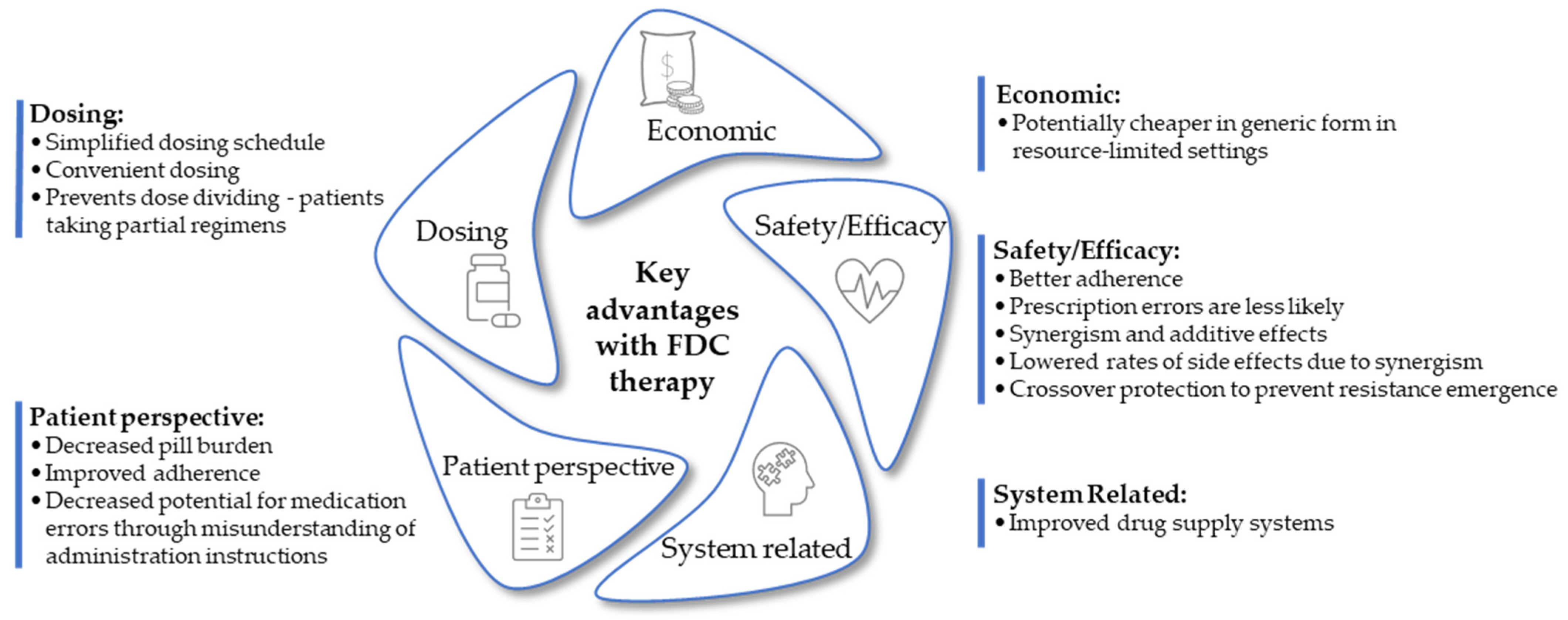
3. Advantages of Fixed-Dose Combination Formulations in Drug Therapy
3.1. Patient Adherence
3.2. Simplified Dosing Schedule
3.3. Prevention of Dose Dividing
3.4. Drug Resistance Prevention
3.5. Synergism
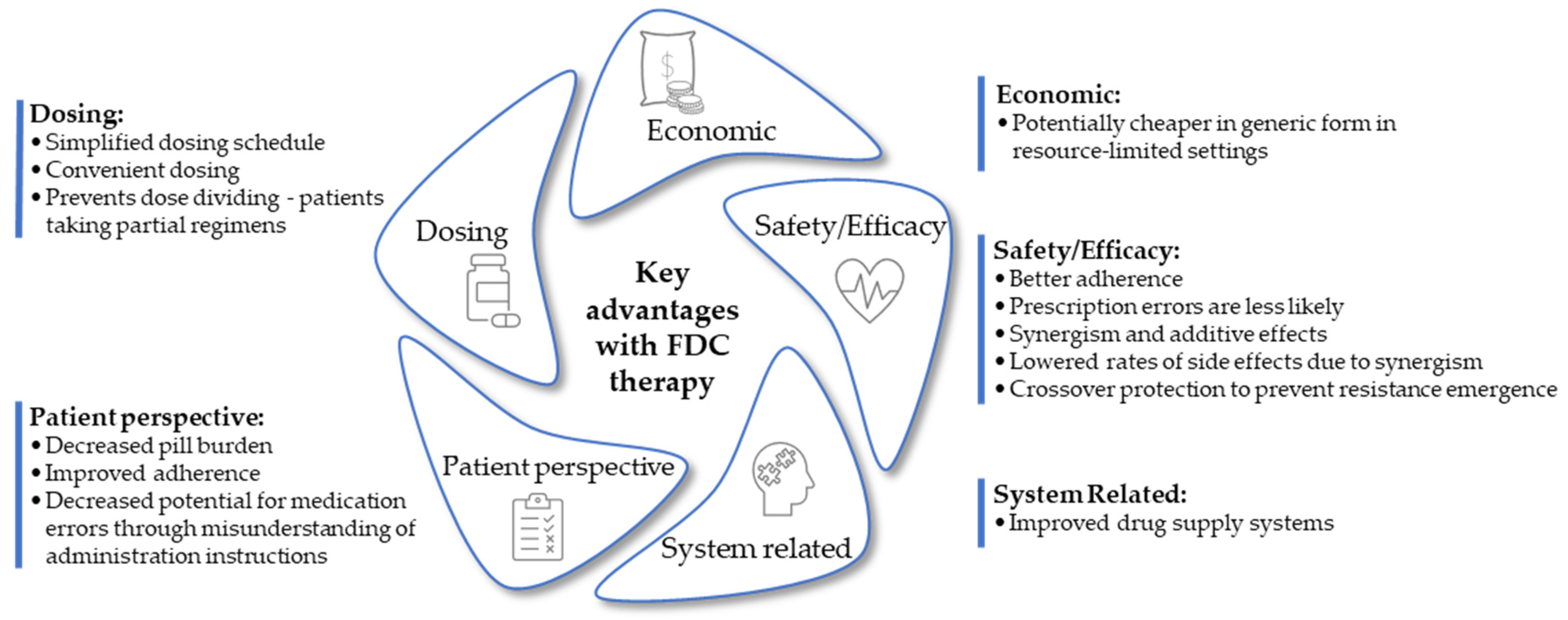
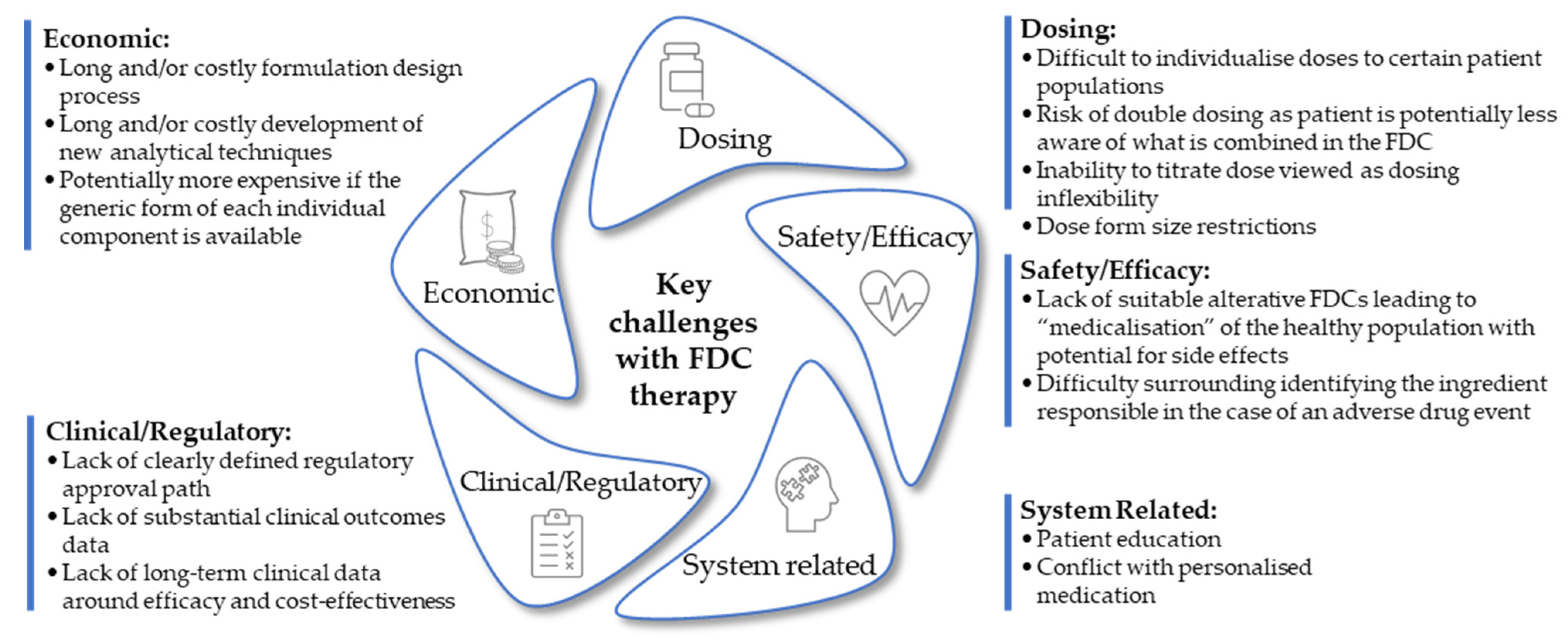
4. Challenges of Fixed-Dose Combination Formulations in Drug Therapy
4.1. Time-Dependent Patient Compliance
4.2. Dose Inflexibility
4.3. Development of Analytical Methods
4.4. Drug Interactions
4.5. Fixed-Dose Combination Therapy Conflict with Personalized Medicine
4.6. Individual Drug Patents Hinder Development of Fixed-Dose Combination Products
5. Conditions Commonly Treated with Fixed-Dose Combination Formulations
5.1. Cardiovascular Disease
5.2. Antibacterials for Systemic Use
5.3. Malaria
5.4. Tuberculosis
5.5. Human Immunodeficiency Virus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brand Name(s) and Companies | Active Pharmaceutical Ingredients Classification | ||||
|---|---|---|---|---|---|
| NRTIs/NtRTIs | NNRTIs | INSTI | PI | PK Enhancer | |
| AluviaTM (Abbott Laboratories); Kaletra® (AbbVie) | Lopinavir (100; 200 mg) | Ritonavir (25; 50 mg) | |||
| Atripla® (MSD); Atroiza (Mylan); Citenvir (Novagen); Odimune (Cipla); Tribuss™ (Aspen) | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | Efavirenz (600 mg) | |||
| Biktarvy® (Aspen) | Emtricitabine (200 mg) Tenofovir alafenamide (25 mg) | Bictegravir (50 mg) | |||
| Combivir® (GlaxoSmithKline, ViiV); Combozil (HeteroDrugs SA); Duovir (Cipla); Adco-lamivudine and zidovudine; Lamzid (Aspen); Loziv (Novagen Pharma) | Lamivudine (150 mg) Zidovudine (300 mg) | ||||
| Complera® (Aspen), Eviplera® (Gilead) | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | Rilpivirine (27.5 mg) | |||
| Delstrigo® (Merck & Co.) | Lamivudine (300 mg) Tenofovir disoproxil (300 mg) | Doravirine (100 mg) | |||
| Dovato (GlaxoSmithKline) | Lamivudine (300 mg) | Dolutegravir (50 mg) | |||
| Epzicom® (US, ViiV), Kivexa® (GSK, ViiV Healthcare); Dumiva (Mylan) | Abacavir (600 mg) Lamivudine (300 mg) | ||||
| Duovir-N (Cipla); Triomune (Cipla); Sonke LamNevStav; Virtrium (Aspen) | Lamivudine (150 mg) Stavudine (30 mg) | Nevirapine (200 mg) | |||
| Genvoya® (Aspen) | Emtricitabine (200 mg) Tenofovir alafenamide (10 mg) | Elvitegravir (150 mg) | Cobicistat (150 mg) | ||
| Juluca (GlaxoSmithKline) | Rilpivirine (25 mg) | Dolutegravir (50 mg) | |||
| Odefsey® (Aspen) | Emtricitabine (200 mg) Tenofovir alafenamide (25 mg) | Rilpivirine (25 mg) | |||
| Stribild® (Gilead) | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | Elvitegravir (150 mg) | Cobicistat (150 mg) | ||
| Symfi® (Mylan); Tenarenz (Aspen) | Lamivudine (300 mg) Tenofovir disoproxil (300 mg) | Efavirenz (600 mg) | |||
| Symtuza® (Janssen Pharmaceutica) | Emtricitabine (200 mg) Tenofovir alafenamide (11.2 mg) | Darunavir (800 mg) | Cobicistat (150 mg) | ||
| Triplavar (Cipla) | Lamivudine (150 mg) Zidovudine (300 mg) | Nevirapine (200 mg) | |||
| Triumeq® (GlaxoSmithKline) | Abacavir (600 mg) Lamivudine (300 mg) | Dolutegravir (50 mg) | |||
| Trizivir® (GlaxoSmithKline) | Abacavir (300 mg) Lamivudine (150 mg) Zidovudine (300 mg) | ||||
| Truvada® (Gilead); Adco-Emtevir (Adcock Ingrams); Tencitab (Aspen); Didivir (Cipla); Tyricten (Aurobindo); Tenemine (Mylan) | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | ||||
5.6. Diabetes Mellitus
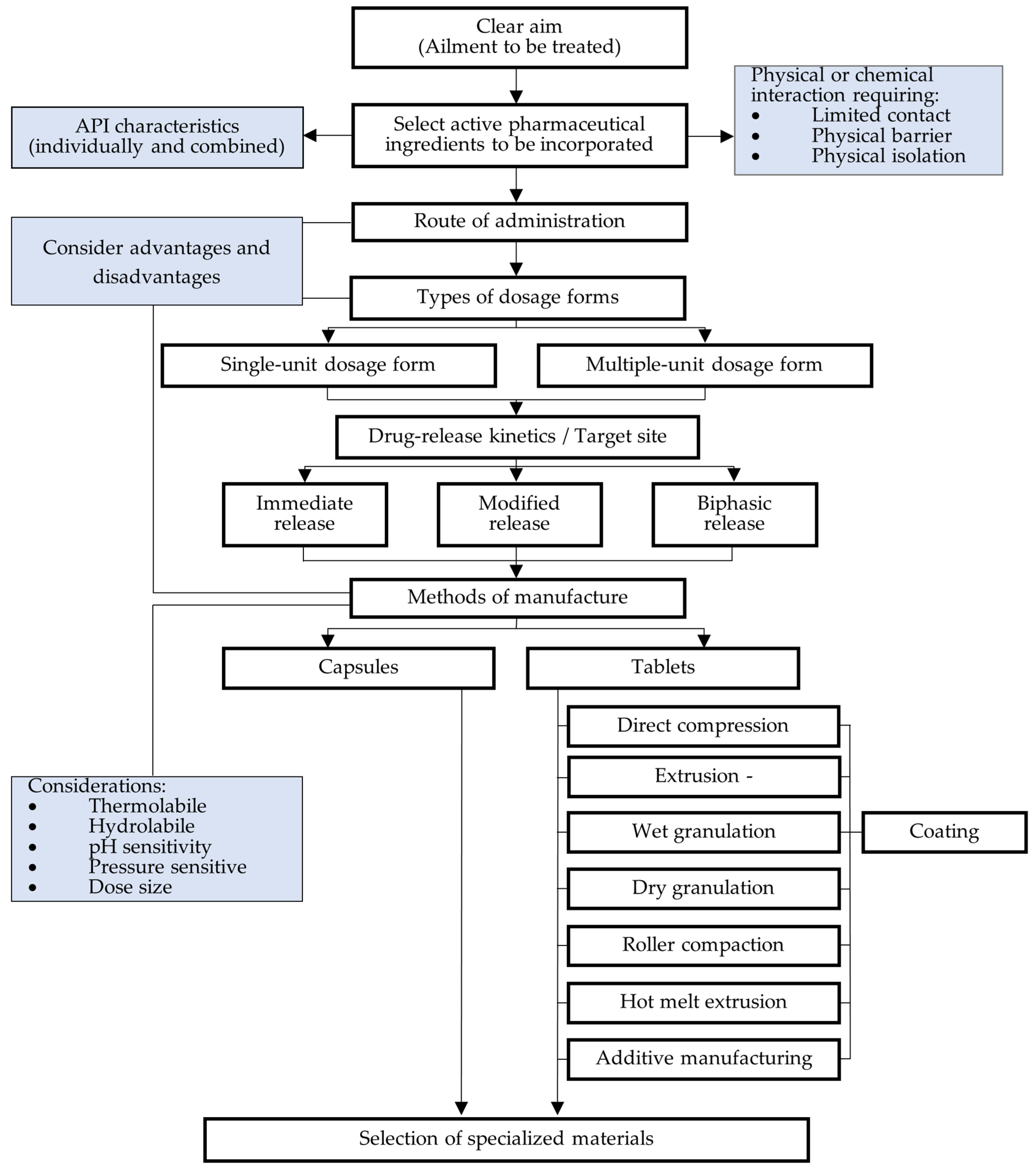
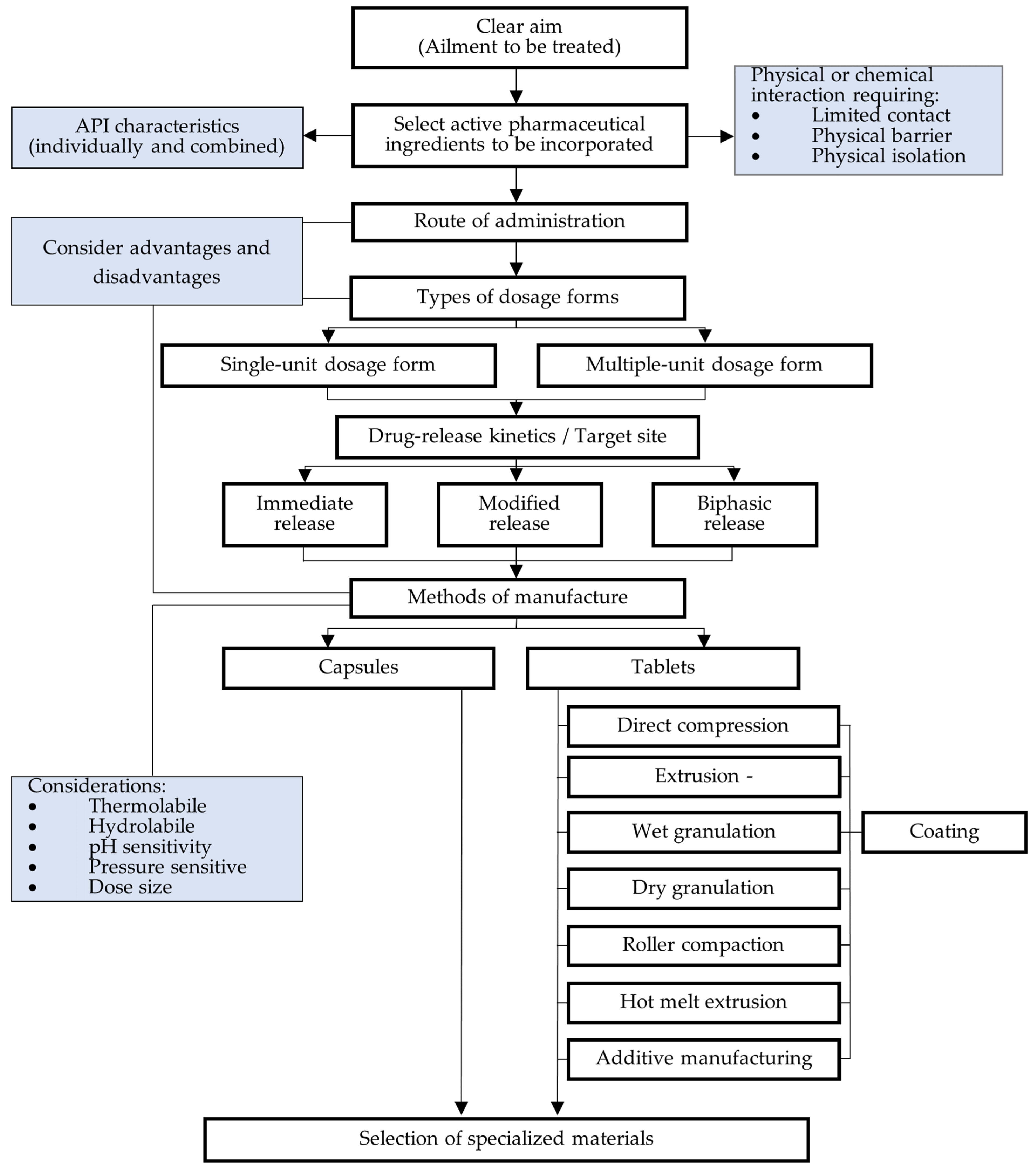
6. Fixed-Dose Combination Formulation Factors for Consideration
7. Formulation Approaches
7.1. Immediate-Release Fixed-Dose Combination Dosage Forms
7.2. Modified-Release Fixed-Dose Combination Dosage Forms
7.2.1. Extended-Release Dosage Forms
Matrix-Type Drug Delivery Systems
Multiple-Unit Pellet Systems
7.2.2. Coating
7.2.3. Delayed-Release Dosage Forms
7.2.4. pH-Responsive Drug Delivery Systems
7.3. Layered Tablets
7.4. Lipid-Based Formulations
7.5. Additive Manufacturing
7.6. Multiple-Unit Delivery Systems
7.7. Layered Tablets with Drug-Free Layers
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wiley, B.; Fuster, V. The concept of the polypill in the prevention of cardiovascular disease. Ann. Glob. Health 2014, 80, 24–34. [Google Scholar] [CrossRef]
- Roser, M.; Ritchie, H.; Spooner, F. Burden of Disease. Available online: https://ourworldindata.org/burden-of-disease (accessed on 17 November 2023).
- WHO. World Health Organization: The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death#:~:text=The%20top%20global%20causes%20of%20death%2C%20in%20order,neonatal%20sepsis%20and%20infections%2C%20and%20preterm%20birth%20complications (accessed on 15 August 2023).
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Albanna, A.S.; Smith, B.M.; Cowan, D.; Menzies, D. Fixed-dose combination antituberculosis therapy: A systematic review and meta-analysis. Eur. Respir. J. 2013, 42, 721–732. [Google Scholar] [CrossRef]
- Gallardo, C.; Comas, D.; Rodríguez, A.; Roqué i Figuls, M.; Parker, L.; Caylà, J.; Bonfill, X. Fixed-dose combinations of drugs versus single-drug formulations for treating pulmonary tuberculosis. Cochrane Database Syst. Rev. 2016, 5, 1–42. [Google Scholar] [CrossRef]
- Subashini, R.; Murugan, A.C.; Nidhina, R. Effect of Ascorbic Acid on dissolution stability of Rifampicin in market fixed dose combination products for Tuberculosis. PharmaTutor 2017, 5, 48–53. [Google Scholar]
- Premji, Z.G. Coartem: The journey to the clinic. Malar. J. 2009, 8 (Suppl. S1), S3. [Google Scholar] [CrossRef]
- Schmieder, R.E.; Ruilope, L.M. Blood pressure control in patients with comorbidities. J. Clin. Hypertens. 2008, 10, 624–631. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Health Organisation. Guidelines for Treatment of Drug-Susceptible Tuberculosis and Patient Care: 2017 Update. Available online: https://apps.who.int/iris/bitstream/handle/10665/255052/9789241550000-eng.pdf;jsessionid=794C08EFC3CD44841D1BF9A4D88E6391?sequence=1 (accessed on 16 November 2023).
- Wald, N.J.; Wald, D.S. The polypill concept. Postgrad. Med. J. 2010, 86, 257–260. [Google Scholar] [CrossRef]
- Wald, D.S.; Wald, N.J. The Polypill in the prevention of cardiovascular disease. Prev. Med. 2011, 52, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Wald, N.J.; Law, M.R. A strategy to reduce cardiovascular disease by more than 80%. BMJ 2003, 326, 1419. [Google Scholar] [CrossRef] [PubMed]
- Janczura, M.; Sip, S.; Cielecka-Piontek, J. The Development of Innovative Dosage Forms of the Fixed-Dose Combination of Active Pharmaceutical Ingredients. Pharmaceutics 2022, 14, 834. [Google Scholar] [CrossRef]
- Sanz, G.; Fuster, V. Polypills for cardiovascular prevention: A step forward? Nat. Rev. Cardiol. 2013, 10, 683–684. [Google Scholar] [CrossRef]
- Webster, R.; Patel, A.; Selak, V.; Billot, L.; Bots, M.L.; Brown, A.; Bullen, C.; Cass, A.; Crengle, S.; Raina Elley, C.; et al. Effectiveness of fixed dose combination medication (‘polypills’) compared with usual care in patients with cardiovascular disease or at high risk: A prospective, individual patient data meta-analysis of 3140 patients in six countries. Int. J. Cardiol. 2016, 205, 147–156. [Google Scholar] [CrossRef] [PubMed]
- WHO. Adherence to Long-Term Therapies: Evidence for Action. Available online: https://iris.who.int/bitstream/handle/10665/42682/9241545992.pdf?sequence=1 (accessed on 7 September 2023).
- DiMatteo, M.R. Variations in patients’ adherence to medical recommendations: A quantitative review of 50 years of research. Med. Care 2004, 42, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V. An alarming threat to secondary prevention: Low compliance (lifestyle) and poor adherence (drugs). Rev. Esp. Cardiol. 2012, 65 (Suppl. S2), 10–16. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Cai, C.; Ai-Fang, Y.; Feng-Min, T.; Li, C.; Bin, W. The effect of placebo adherence on reducing cardiovascular mortality: A meta-analysis. Clin. Res. Cardiol. 2014, 103, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Bitton, A.; Choudhry, N.K.; Matlin, O.S.; Swanton, K.; Shrank, W.H. The impact of medication adherence on coronary artery disease costs and outcomes: A systematic review. Am. J. Med. 2013, 126, 357.e7. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Islam, S.; Chow, C.K.; Rangarajan, S.; Dagenais, G.; Diaz, R.; Gupta, R.; Kelishadi, R.; Iqbal, R.; Avezum, A.; et al. Use of secondary prevention drugs for cardiovascular disease in the community in high-income, middle-income, and low-income countries (the PURE Study): A prospective epidemiological survey. Lancet 2011, 378, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Hugtenburg, J.G.; Timmers, L.; Elders, P.J.; Vervloet, M.; van Dijk, L. Definitions, variants, and causes of nonadherence with medication: A challenge for tailored interventions. Patient Prefer. Adherence 2013, 7, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Copeland-Halperin, R.; Fuster, V. Aiming at strategies for a complex problem of medical nonadherence. Glob. Heart 2013, 8, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Castellano, J.M.; Onuma, O.K. Putting polypills into practice: Challenges and lessons learned. Lancet 2017, 389, 1066–1074. [Google Scholar] [CrossRef]
- Pan, F.; Chernew, M.E.; Fendrick, A.M. Impact of fixed-dose combination drugs on adherence to prescription medications. J. Gen. Intern. Med. 2008, 23, 611–614. [Google Scholar] [CrossRef]
- Arya, D.S.; Chowdhury, S.; Chawla, R.; Das, A.K.; Ganie, M.A.; Kumar, K.M.P.; Nadkar, M.Y.; Rajput, R. Clinical Benefits of Fixed Dose Combinations Translated to Improved Patient Compliance. J. Assoc. Physicians India 2019, 67, 58–64. [Google Scholar]
- SAMF. South African Medicine Formulary, 14th ed.; Health and Medical Publishing Group (PTY) Ltd.: Rondebosch, South Africa, 2022. [Google Scholar]
- Tsiligiannis, A.; Sfouni, M.; Nalda-Molina, R.; Dokoumetzidis, A. Optimization of a paediatric fixed dose combination mini-tablet and dosing regimen for the first line treatment of tuberculosis. Eur. J. Pharm. Sci. 2019, 138, 105016. [Google Scholar] [CrossRef]
- Song, S.; Kim, S.; Shin, S.; Lee, Y.; Lee, E. Evaluation of Prescription Medication Sharing Among Adults in South Korea: A Cross-Sectional Survey. Front. Pharmacol. 2022, 13, 773454. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, D.A. How drug resistance emerges as a result of poor compliance during short course chemotherapy for tuberculosis. Int. J. Tuberc. Lung Dis. 1998, 2, 10–15. [Google Scholar]
- Hussein, W. Fixed-Dose Combination in Diabetes Management. Safe Smart 3D 2020, 1–20. [Google Scholar]
- Sica, D.A. Rationale for fixed-dose combinations in the treatment of hypertension: The cycle repeats. Drugs 2002, 62, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Godman, B.; McCabe, H.; DLeong, T.; Mueller, D.; Martin, A.P.; Hoxha, I.; Mwita, J.C.; Rwegerera, G.M.; Massele, A.; Costa, J.D.O.; et al. Fixed dose drug combinations—Are they pharmacoeconomically sound? Findings and implications especially for lower- and middle-income countries. Expert Rev. Pharmacoeconomics Outcomes Res. 2020, 20, 1–26. [Google Scholar] [CrossRef]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef] [PubMed]
- Omollo, C.; Singh, V.; Kigondu, E.; Wasuna, A.; Agarwal, P.; Moosa, A.; Ioerger, T.R.; Mizrahi, V.; Chibale, K.; Warner, D.F. Developing synergistic drug combinations to restore antibiotic sensitivity in drug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2023, 65. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Naik, N.; Srinath Reddy, K. Strengths and Limitations of Using the Polypill in Cardiovascular Prevention. Curr. Cardiol. Rep. 2017, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.S. Combine and conquer: Advantages and disadvantages of fixed-dose combination therapy. Diabetes Obes. Metab. 2013, 15, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, A.; Drame, K.; Geutjens, S.; Airaksinen, M. Does the Polypill Improve Patient Adherence Compared to Its Individual Formulations? A Systematic Review. Pharmaceutics 2020, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Sanz, G.; Peñalvo, J.L.; Bansilal, S.; Fernández-Ortiz, A.; Alvarez, L.; Guzmán, L.; Linares, J.C.; García, F.; D’Aniello, F.; et al. A Polypill Strategy to Improve Adherence: Results from the FOCUS Project. J. Am. Coll. Cardiol. 2014, 64, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Laba, T.L.; Howard, K.; Rose, J.; Peiris, D.; Redfern, J.; Usherwood, T.; Cass, A.; Patel, A.; Jan, S. Patient Preferences for a Polypill for the Prevention of Cardiovascular Diseases. Ann. Pharmacother. 2015, 49, 528–539. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.; Lu, M.; Stämpfli, A. Simultaneous determination of elbasvir and grazoprevir in fixed-dose combination and mass spectral characterization of each degradation product by UHPLC-ESI-QTOF-MS/MS. J. Pharm. Biomed. Anal. 2020, 178, 112964. [Google Scholar] [CrossRef]
- Kurmi, M.; Jayaraman, K.; Natarajan, S.; Kumar, G.S.; Bhutani, H.; Bajpai, L. Rapid and efficient chiral method development for lamivudine and tenofovir disoproxil fumarate fixed dose combination using ultra-high performance supercritical fluid chromatography: A design of experiment approach. J. Chromatogr. A 2020, 1625, 461257. [Google Scholar] [CrossRef]
- Li, Z. Chiral Separation of Lamivudine by Capillary Zone Electrophoresis. Asian J. Chem. 2013, 25, 7847–7852. [Google Scholar] [CrossRef]
- Seshachalam, U.; Rajababu, B.; Haribabu, B.; Chandrasekhar, K.B. Enantiomeric Separation of Tenofovir on an Achiral C18 Column by HPLC Using L-Phenylalanine as a Chiral Mobile Phase Additive. J. Liq. Chromatogr. Relat. Technol. 2007, 31, 410–420. [Google Scholar] [CrossRef]
- Heydari, R.; Shamsipur, M. Enantiomeric Separation and Quantitation of Tenofovir Disoproxil Fumarate Using Amylose-Based Chiral Stationary Phases by High-Performance Liquid Chromatography. Acta Chromatogr. 2015, 27, 583–595. [Google Scholar] [CrossRef]
- Subbarao, M.; Ramakrishna, M.; Mogilireddy, D. Chiral seperation of lamivudine enantiomer by HPLC using cellulose tris (3,5-Dichlorophenylcarbamate) as a chiral stationary phase. Indo Am. J. Pharm. Res. 2016, 6, 5487–5493. [Google Scholar]
- Bhutani, H.; Mariappan, T.T.; Singh, S. The physical and chemical stability of anti-tuberculosis fixed-dose combination products under accelerated climatic conditions. Int. J. Tuberc. Lung Dis. 2004, 8, 1073–1080. [Google Scholar] [PubMed]
- Bhutani, H.; Singh, S.; Jindal, K.C.; Chakraborti, A.K. Mechanistic explanation to the catalysis by pyrazinamide and ethambutol of reaction between rifampicin and isoniazid in anti-TB FDCs. J. Pharm. Biomed. Anal. 2005, 39, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Shishoo, C.J.; Shah, S.A.; Rathod, I.S.; Savale, S.S.; Vora, M.J. Impaired bioavailability of rifampicin in presence of isoniazid from fixed dose combination (FDC) formulation. Int. J. Pharm. 2001, 228, 53–67. [Google Scholar] [CrossRef]
- Singh, S.; Mariappan, T.T.; Sharda, N.; Singh, B. Degradation of Rifampicin, Isoniazid and Pyrazinamide from Prepared Mixtures and Marketed Single and Combination Products Under Acid Conditions. Pharm. Pharmacol. Commun. 2000, 6, 491–494. [Google Scholar] [CrossRef]
- Fravel, M.A.; Ernst, M. Drug Interactions with Antihypertensives. Curr. Hypertens. Rep. 2021, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Elliott, W.J. Drug interactions and drugs that affect blood pressure. J. Clin. Hypertens. 2006, 8, 731–737. [Google Scholar] [CrossRef]
- Cooper-DeHoff, R.; Bird, S.; Nichols, G.; Delaney, J.; Winterstein, A. Antihypertensive Drug Class Interactions and Risk for Incident Diabetes: A Nested Case–Control Study. J. Am. Heart Assoc. 2013, 2, e000125. [Google Scholar] [CrossRef]
- Oyewumi, M. 3D Printing Technology in Pharmaceutical Drug Delivery: Prospects and Challenges. J. Biomol. Res. Ther. 2015, 4. [Google Scholar] [CrossRef]
- Jakka, S.; Rossbach, M. An economic perspective on personalized medicine. Hugo J. 2013, 7, 1. [Google Scholar] [CrossRef]
- Goetz, L.H.; Schork, N.J. Personalized medicine: Motivation, challenges, and progress. Fertil. Steril. 2018, 109, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Rodriguez-Monguio, R.; Seoane-Vazquez, E. Fixed-Dose Combination Drug Approvals, Patents and Market Exclusivities Compared to Single Active Ingredient Pharmaceuticals. PLoS ONE 2015, 10, e0140708. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Health Organization Model List of Essential Medications. Available online: https://list.essentialmeds.org/ (accessed on 15 September 2023).
- Waeber, B.; Ruilope, L.M. Amlodipine and valsartan as components of a rational and effective fixed-dose combination. Vasc. Health Risk Manag. 2009, 5, 165–174. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline on Clinical Development of Fixed Combination Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-development-fixed-combination-medicinal-products-revision-2_en.pdf (accessed on 11 June 2023).
- DiPette, D.J.; Skeete, J.; Ridley, E.; Campbell, N.R.C.; Lopez-Jaramillo, P.; Kishore, S.P.; Jaffe, M.G.; Coca, A.; Townsend, R.R.; Ordunez, P. Fixed-dose combination pharmacologic therapy to improve hypertension control worldwide: Clinical perspective and policy implications. J. Clin. Hypertens. 2019, 21, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Gradman, A.H.; Basile, J.N.; Carter, B.L.; Bakris, G.L.; Materson, B.J.; Black, H.R.; Izzo, J.L., Jr.; Oparil, S.; Weber, M.A. Combination therapy in hypertension. J. Am. Soc. Hypertens. 2010, 4, 90–98. [Google Scholar] [CrossRef]
- Nansseu, J.R.; Tankeu, A.T.; Kamtchum-Tatuene, J.; Noubiap, J.J. Fixed-dose combination therapy to reduce the growing burden of cardiovascular disease in low- and middle-income countries: Feasibility and challenges. J. Clin. Hypertens. 2018, 20, 168–173. [Google Scholar] [CrossRef] [PubMed]
- WHO. Guidelines on Post-Exposure Prophylaxis for HIV and the Use of Co-Trimoxazole Prophylaxis for HIV-Related Infections among Adults, Adolescents and Children; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Vliegenthart-Jongbloed, K.; Jacobs, J. Not recommended fixed-dose antibiotic combinations in low- and middle-income countries–the example of Tanzania. Antimicrob. Resist. Infect. Control 2023, 12, 37. [Google Scholar] [CrossRef]
- Wushouer, H.; Hu, L.; Zhou, Y.; Yang, Y.; Du, K.; Deng, Y.; Yan, Q.; Yang, X.; Chen, Z.; Zheng, B.; et al. Trends of Fixed-Dose Combination Antibiotic Consumption in Hospitals in China: Analysis of Data from the Center for Antibacterial Surveillance, 2013–2019. Antibiotics 2022, 11, 957. [Google Scholar] [CrossRef]
- WHO. Recommended Comparator Products: Anti-Malarial Medicines Guidance Document. Available online: https://extranet.who.int/prequal/sites/default/files/document_files/Comparator_MA_31October2023.pdf (accessed on 23 August 2023).
- EMA. Pyramax: Opinion on Medicine for Use Outside EU. Available online: https://www.ema.europa.eu/en/opinion-medicine-use-outside-EU/human/pyramax (accessed on 11 September 2023).
- Patil, C.; Katare, S.; Baig, M.; Doifode, S. Fixed dose combination of arterolane and piperaquine: A newer prospect in antimalarial therapy. Ann. Med. Health Sci. Res. 2014, 4, 466–471. [Google Scholar] [CrossRef]
- Scholar, E. Lumefantrine. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. [Google Scholar]
- Prabhu, P.; Suryavanshi, S.; Pathak, S.; Sharma, S.; Patravale, V. Artemether-lumefantrine nanostructured lipid carriers for oral malaria therapy: Enhanced efficacy at reduced dose and dosing frequency. Int. J. Pharm. 2016, 511, 473–487. [Google Scholar] [CrossRef]
- Nosten, F.; White, N.J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007, 77, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Leonardi, A.; Cupri, S.; Puglisi, G.; Pignatello, R. Pharmaceutical and biomedical applications of lipid-based nanocarriers. Pharm. Pat. Anal. 2014, 3, 199–215. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, F.; Liu, W.; Wan, J.; Wan, C.; Xu, H.; Lam, C.W.; Yang, X. Nanostructured lipid carriers as a novel oral delivery system for triptolide: Induced changes in pharmacokinetics profile associated with reduced toxicity in male rats. Int. J. Nanomed. 2014, 9, 1049–1063. [Google Scholar] [CrossRef]
- Uzondu, S.; Echezona, A.; Nwagwu, C.; Onugwu, A.; Ugorji, O.; Agbo, C.; Kenechukwu, F.; Ogbonna, J.; Akpa, P.; Nnamani, P.; et al. Combating Antimalarial Drug Resistance: Recent Advances and Future Perspectives. In Malaria–Recent Advances and New Perspectives; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Marwa, K.; Kapesa, A.; Baraka, V.; Konje, E.; Kidenya, B.; Mukonzo, J.; Kamugisha, E.; Swedberg, G. Therapeutic efficacy of artemether-lumefantrine, artesunate-amodiaquine and dihydroartemisinin-piperaquine in the treatment of uncomplicated Plasmodium falciparum malaria in Sub-Saharan Africa: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0264339. [Google Scholar] [CrossRef] [PubMed]
- Ezzet, F.; van Vugt, M.; Nosten, F.; Looareesuwan, S.; White, N.J. Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria. Antimicrob. Agents Chemother. 2000, 44, 697–704. [Google Scholar] [CrossRef]
- Ogutu, B.; Yeka, A.; Kusemererwa, S.; Thompson, R.; Tinto, H.; Toure, A.O.; Uthaisin, C.; Verma, A.; Kibuuka, A.; Lingani, M.; et al. Ganaplacide (KAF156) plus lumefantrine solid dispersion formulation combination for uncomplicated Plasmodium falciparum malaria: An open-label, multicentre, parallel-group, randomised, controlled, phase 2 trial. Lancet 2023, 23, 1051–1061. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Efficacy and Safety of KAF156 in Combination with LUM-SDF in Adults and Children with Uncomplicated Plasmodium Falciparum Malaria. Available online: https://clinicaltrials.gov/study/NCT03167242 (accessed on 8 December 2023).
- Blomberg, B.; Spinaci, S.; Fourie, B.; Laing, R. The rationale for recommending fixed-dose combination tablets for treatment of tuberculosis. Bull. World Health Organ. 2001, 79, 61–68. [Google Scholar]
- McIlleron, H.; Wash, P.; Burger, A.; Folb, P.; Smith, P. Widespread distribution of a single drug rifampicin formulation of inferior bioavailability in South Africa. Int. J. Tuberc. Lung Dis. 2002, 6, 356–361. [Google Scholar]
- Rajaram, S.; Vemuri, V.D.; Natham, R. Ascorbic acid improves stability and pharmacokinetics of rifampicin in the presence of isoniazid. J. Pharm. Biomed. Anal. 2014, 100, 103–108. [Google Scholar] [CrossRef]
- Jönsson, S.; Karlsson, M.O. A rational approach for selection of optimal covariate-based dosing strategies. Clin. Pharmacol. Ther. 2003, 73, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Vinks, A.A.; Emoto, C.; Fukuda, T. Modeling and simulation in pediatric drug therapy: Application of pharmacometrics to define the right dose for children. Clin. Pharmacol. Ther. 2015, 98, 298–308. [Google Scholar] [CrossRef]
- Meyers, T.; Moultrie, H.; Naidoo, K.; Cotton, M.; Eley, B.; Sherman, G. Challenges to Pediatric HIV Care and Treatment in South Africa. J. Infect. Dis. 2007, 196, S474–S481. [Google Scholar] [CrossRef]
- Hirasen, K.; Evans, D.; Maskew, M.; Sanne, I.M.; Shearer, K.; Govathson, C.; Malete, G.; Kluberg, S.A.; Fox, M.P. The right combination-treatment outcomes among HIV-positive patients initiating first-line fixed-dose antiretroviral therapy in a public sector HIV clinic in Johannesburg, South Africa. Clin. Epidemiol. 2018, 10, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Lopez, F.L.; Ernest, T.B.; Tuleu, C.; Gul, M.O. Formulation approaches to pediatric oral drug delivery: Benefits and limitations of current platforms. Expert. Opin. Drug Deliv. 2015, 12, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Ivanovska, V.; Rademaker, C.M.; van Dijk, L.; Mantel-Teeuwisse, A.K. Pediatric drug formulations: A review of challenges and progress. Pediatrics 2014, 134, 361–372. [Google Scholar] [CrossRef]
- Cella, M.; Knibbe, C.; Danhof, M.; Della Pasqua, O. What is the right dose for children? Br. J. Clin. Pharmacol. 2010, 70, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J.; Holford, N.H. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 303–332. [Google Scholar] [CrossRef]
- Liu, T.; Ghafoori, P.; Gobburu, J.V. Allometry Is a Reasonable Choice in Pediatric Drug Development. J. Clin. Pharmacol. 2017, 57, 469–475. [Google Scholar] [CrossRef]
- Mahmood, I. Prediction of drug clearance in children from adults: A comparison of several allometric methods. Br. J. Clin. Pharmacol. 2006, 61, 545–557. [Google Scholar] [CrossRef]
- Dorlo, T.P.; Huitema, A.D.; Beijnen, J.H.; de Vries, P.J. Optimal dosing of miltefosine in children and adults with visceral leishmaniasis. Antimicrob. Agents Chemother. 2012, 56, 3864–3872. [Google Scholar] [CrossRef]
- Tarning, J.; Zongo, I.; Somé, F.A.; Rouamba, N.; Parikh, S.; Rosenthal, P.J.; Hanpithakpong, W.; Jongrak, N.; Day, N.P.J.; White, N.J.; et al. Population Pharmacokinetics and Pharmacodynamics of Piperaquine in Children with Uncomplicated Falciparum Malaria. Clin. Pharmacol. Ther. 2012, 91, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.J.; Driscoll, T.A.; Milligan, P.A.; Wood, N.D.; Schlamm, H.T.; Groll, A.H.; Jafri, H.S.; Arrieta, A.C.; Klein, N.; Lutsar, I. Pharmacokinetics, Safety, and Tolerability of Voriconazole in Immunocompromised Children. Antimicrob. Agents Chemother. 2010, 54, 4116–4123. [Google Scholar] [CrossRef] [PubMed]
- Fillekes, Q.; Natukunda, E.; Balungi, J.; Kendall, L.; Bwakura-Dangarembizi, M.; Keishanyu, R.; Ferrier, A.; Lutakome, J.; Gibb, D.M.; Burger, D.M.; et al. Pediatric underdosing of efavirenz: A pharmacokinetic study in Uganda. J. Acquir. Immune Defic. Syndr. 2011, 58, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Rostami-Hodjegan, A.; Tucker, G.T. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 2006, 45, 931–956. [Google Scholar] [CrossRef] [PubMed]
- HIV.gov, C.i. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Available online: https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/what-start-initial-combination-regimens-antiretroviral-naive-1 (accessed on 5 October 2023).
- HIV.gov. Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection. Available online: https://clinicalinfo.hiv.gov/en/guidelines/pediatric-arv/appendix-a-table-2-antiretroviral-fixed-dose-combination-tablets-and-copackaged-formulations (accessed on 15 November 2023).
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycemia in type 2 diabetes, 2015: A patient-centered approach: Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015, 38, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Das, A.K.; Priya, G.; Ghosh, S.; Mehrotra, R.N.; Das, S.; Shah, P.; Bajaj, S.; Deshmukh, V.; Sanyal, D.; et al. Fixed-dose combination in management of type 2 diabetes mellitus: Expert opinion from an international panel. J. Fam. Med. Prim. Care 2020, 9, 5450–5457. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Huang, C.N.; Hung, Y.J.; Kwok, C.F.; Sun, J.H.; Pei, D.; Yang, C.Y.; Chen, C.C.; Lin, C.L.; Sheu, W.H. Acarbose plus metformin fixed-dose combination outperforms acarbose monotherapy for type 2 diabetes. Diabetes Res. Clin. Pract. 2013, 102, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Brown, A.; Fischer, J.; Jain, A.; Littlejohn, T.; Nadeau, D.; Sussman, A.; Taylor, T.; Krol, A.; Magner, J. Efficacy and Safety of Acarbose in Metformin-Treated Patients with Type 2 Diabetes. Diabetes Care 1998, 21, 2050–2055. [Google Scholar] [CrossRef]
- Phillips, P.; Karrasch, J.; Scott, R.; Wilson, D.; Moses, R. Acarbose improves glycemic control in overweight type 2 diabetic patients insufficiently treated with metformin. Diabetes Care 2003, 26, 269–273. [Google Scholar] [CrossRef]
- Halimi, S.; Le Berre, M.A.; Grangé, V. Efficacy and safety of acarbose add-on therapy in the treatment of overweight patients with Type 2 diabetes inadequately controlled with metformin: A double-blind, placebo-controlled study. Diabetes Res. Clin. Pract. 2000, 50, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Lin, S.D.; Lee, W.J.; Su, S.L.; Lee, I.T.; Tu, S.T.; Tseng, Y.H.; Lin, S.Y.; Sheu, W.H. Effects of acarbose versus glibenclamide on glycemic excursion and oxidative stress in type 2 diabetic patients inadequately controlled by metformin: A 24-week, randomized, open-label, parallel-group comparison. Clin. Ther. 2011, 33, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.C.; Cheng, H.T.; Stables, C.L.; Smith, A.L.; Feldman, E.L. Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Hammad, M.A.; Syed Sulaiman, S.A.; Alghamdi, S.; Mangi, A.A.; Aziz, N.A.; Mohamed Noor, D.A. Statins-related peripheral neuropathy among diabetic patients. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.M.M.; Chua, Z.J.Y.; Tan, J.C.; Yang, Y.; Liao, Z.; Zhao, Y. From Pre-Diabetes to Diabetes: Diagnosis, Treatments and Translational Research. Medicina 2019, 55, 546. [Google Scholar] [CrossRef]
- McConnell, E.L.; Basit, A.W. Aulton’s Pharmaceutics: The Design and Manufacture of Medicines, 4th ed.; Churchill Livingstone Elsevier: London, UK, 2013. [Google Scholar]
- Svensson, E.M.; Yngman, G.; Denti, P.; McIlleron, H.; Kjellsson, M.C.; Karlsson, M.O. Evidence-Based Design of Fixed-Dose Combinations: Principles and Application to Pediatric Anti-Tuberculosis Therapy. Clin. Pharmacokinet. 2018, 57, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Nart, V.; Beringhs, A.O.; França, M.T.; de Espíndola, B.; Pezzini, B.R.; Stulzer, H.K. Carnauba wax as a promising excipient in melt granulation targeting the preparation of mini-tablets for sustained release of highly soluble drugs. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 70, 250–257. [Google Scholar] [CrossRef]
- Smith, D.M.; Kapoor, Y.; Klinzing, G.R.; Procopio, A.T. Pharmaceutical 3D printing: Design and qualification of a single step print and fill capsule. Int. J. Pharm. 2018, 544, 21–30. [Google Scholar] [CrossRef]
- Fernández-García, R.; Prada, M.; Bolas-Fernández, F.; Ballesteros, M.; Serrano, D. Oral Fixed-Dose Combination Pharmaceutical Products: Industrial Manufacturing Versus Personalized 3D Printing. Pharm. Res. 2020, 37, 132. [Google Scholar] [CrossRef]
- Pérez, P.; Suñé-Negre, J.M.; Miñarro, M.; Roig, M.; Fuster, R.; García-Montoya, E.; Hernández, C.; Ruhí, R.; Ticó, J.R. A new expert systems (SeDeM diagram) for control batch powder formulation and preformulation drug products. Eur. J. Pharm. Biopharm. 2006, 64, 351–359. [Google Scholar] [CrossRef]
- Singh, I.; Kumar, P. Preformulation studies for direct compression suitability of cefuroxime axetil and paracetamol: A graphical representation using SeDeM diagram. Acta Pol. Pharm. 2012, 69, 87–93. [Google Scholar] [PubMed]
- Suñé-Negre, J.; Garcia, E.; Lozano, P.; Díaz, J.; Carreras, M.; Fuster, R.; Carmona, M.; Ticó, J. SeDeM Diagram: A New Expert System for the Formulation of Drugs in Solid Form. In Vizureanu, Petrică; IntechOpen: London, UK, 2011. [Google Scholar]
- Suñé-Negre, J.M.; Pérez-Lozano, P.; Miñarro, M.; Roig, M.; Fuster, R.; Hernández, C.; Ruhí, R.; García-Montoya, E.; Ticó, J.R. Application of the SeDeM Diagram and a new mathematical equation in the design of direct compression tablet formulation. Eur. J. Pharm. Biopharm. 2008, 69, 1029–1039. [Google Scholar] [CrossRef]
- Suñé-Negre, J.M.; Roig, M.; Fuster, R.; Hernández, C.; Ruhí, R.; García-Montoya, E.; Pérez-Lozano, P.; Miñarro, M.; Ticó, J.R. New classification of directly compressible (DC) excipients in function of the SeDeM Diagarm Expert System. Int. J. Pharm. 2014, 470, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; He, X.; Zhu, L.; Chen, B. Chapter 20-Product and Process Development of Solid Oral Dosage Forms. In Developing Solid Oral Dosage Forms, 2nd ed.; Qiu, Y., Chen, Y., Zhang, G.G.Z., Yu, L., Mantri, R.V., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 555–591. [Google Scholar]
- Alderborn, G. Tablets and compaction. In Aulton’s Pharmaceutics-The Design and Manufacture of Medicines, 3rd ed; Churchill Livingstone: Edinburgh, Scotland, 2007. [Google Scholar]
- Rojas, J.; Ciro, Y.; Correa, L. Functionality of chitin as a direct compression excipient: An acetaminophen comparative study. Carbohydr. Polym. 2014, 103, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Thoorens, G.; Krier, F.; Leclercq, B.; Carlin, B.; Evrard, B. Microcrystalline cellulose, a direct compression binder in a quality by design environment—A review. Int. J. Pharm. 2014, 473, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Lucka, M.; Hanke, T. Sieve Analysis Different Sieving Methods for a Variety of Applications. Available online: https://www.researchgate.net/publication/309011437_Sieve_Analysis_Different_sieving_methods_for_a_variety_of_applications (accessed on 16 November 2023).
- Allen, T. Particle Size Analysis by Sieving. In Powder Sampling and Particle Size Determination; Elsevier: Amsterdam, The Netherlands, 2003; pp. 208–250. [Google Scholar] [CrossRef]
- Kararli, T.T.; Needham, T.E.; Seul, C.J.; Finnegan, P.M. Solid-state interaction of magnesium oxide and ibuprofen to form a salt. Pharm. Res. 1989, 6, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Dedroog, S.; Pas, T.; Vergauwen, B.; Huygens, C.; Van den Mooter, G. Solid-state analysis of amorphous solid dispersions: Why DSC and XRPD may not be regarded as stand-alone techniques. J. Pharm. Biomed. Anal. 2020, 178, 112937. [Google Scholar] [CrossRef]
- Maderuelo, C.; Zarzuelo, A.; Lanao, J.M. Critical factors in the release of drugs from sustained release hydrophilic matrices. J. Control. Release 2011, 154, 2–19. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, T.; Hu, X. Novel polysaccharides-based nanoparticle carriers prepared by polyelectrolyte complexation for protein drug delivery. Polym. Bull. 2012, 68, 1183–1199. [Google Scholar] [CrossRef]
- Xiao, Y.; Xu, W.; Zhu, Q.; Yan, B.; Yang, D.; Yang, J.; He, X.; Liang, S.; Hu, X. Preparation and characterization of a novel pachyman-based pharmaceutical aid. II: A pH-sensitive, biodegradable and biocompatible hydrogel for controlled release of protein drugs. Carbohydr. Polym. 2009, 77, 612–620. [Google Scholar] [CrossRef]
- Ozarde, S.Y.; Sarvi, S.; Polshettiwar, S.A.; Kuchekar, B.S. Multiple-Unit-Pellet System (MUPS): A Novel Approach for Drug Delivery. Drug Invent. Today 2012, 4, 603–609. [Google Scholar]
- Tang, E.S.K.; Chan, L.W.; Heng, P.W.S. Coating of multiparticulates for sustained release. Am. J. Drug Deliv. 2005, 3, 17–28. [Google Scholar] [CrossRef]
- Asghar, L.F.; Chandran, S. Multiparticulate formulation approach to colon specific drug delivery: Current perspectives. J. Pharm. Pharm Sci 2006, 9, 327–338. [Google Scholar] [PubMed]
- Zeeshan, F.; Bukhari, N.I. Development and evaluation of a novel modified-release pellet-based tablet system for the delivery of loratadine and pseudoephedrine hydrochloride as model drugs. AAPS PharmSciTech 2010, 11, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.S.; Majumdar, S.; Rao, M.E.B. Multiparticulate Drug Delivery Systems for Controlled Release. Trop. J. Pharm. Res. 2008, 7, 1067–1075. [Google Scholar] [CrossRef]
- Patel, S.A.; Patel, N.G.; Misra, M.; Joshi, A. Controlled-release domperidone pellets compressed into fast disintegrating tablets forming a multiple-unit pellet system (MUPS). J. Drug Deliv. Sci. Technol. 2018, 45, 220–229. [Google Scholar] [CrossRef]
- Mount, D.L.; Schwartz, J.B. Formulation and Compaction of Nonfracturing Deformable Coated Beads. Drug Dev. Ind. Pharm. 1996, 22, 609–621. [Google Scholar] [CrossRef]
- Bansal, P.; Vasireddy, S.; Plakogiannis, F.; Parikh, D. Effect of compression on the release properties of polymer coated niacin granules. J. Control. Release 1993, 27, 157–163. [Google Scholar] [CrossRef]
- Bodmeier, R. Tableting of coated pellets. Eur. J. Pharm. Biopharm. 1997, 43, 1–8. [Google Scholar] [CrossRef]
- Maganti, L.; Çelik, M. Compaction studies on pellets: II. Coated pellets. Int. J. Pharm. 1994, 103, 55–67. [Google Scholar] [CrossRef]
- Juslin, M.; Turakka, L.; Puumalainen, P. Controlled release tablets. Part 1: The use of pellets coated with a retarding acrylate plastic in tabletting. Pharm. Ind. 1980, 42, 829–832. [Google Scholar]
- Beckert, T.E.; Lehmann, K.; Schmidt, P.C. Compression of enteric-coated pellets to disintegrating tablets: Uniformity of dosage units. Powder Technol. 1998, 96, 248–254. [Google Scholar] [CrossRef]
- Clelik, M.; Maganti, L. Formulation and Compaction of Microspheres. Drug Dev. Ind. Pharm. 1994, 20, 3151–3173. [Google Scholar] [CrossRef]
- Chen, T.; Li, J.; Chen, T.; Sun, C.C.; Zheng, Y. Tablets of multi-unit pellet system for controlled drug delivery. J. Control. Release 2017, 262, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Nicklasson, F.; Alderborn, G. Effect of pellet size on degree of deformation and densification during compression and on compactability of microcrystalline cellulose pellets. Int. J. Pharm. 1998, 163, 35–48. [Google Scholar] [CrossRef]
- Johansson, B.; Wikberg, M.; Ek, R.; Alderborn, G. Compression behaviour and compactability of microcrystalline cellulose pellets in relationship to their pore structure and mechanical properties. Int. J. Pharm. 1995, 117, 57–73. [Google Scholar] [CrossRef]
- SAHPRA. Professional Information for Lopimune 40/10 Oral Pellets. Available online: https://pi-pil-repository.sahpra.org.za/wp-content/uploads/2023/02/pi-lopimune.pdf (accessed on 11 November 2023).
- Wilson, D.I.; Rough, S.L. Chapter 3 Extrusion—Spheronisation. In Handbook of Powder Technology; Salman, A.D., Hounslow, M.J., Seville, J.P.K., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2007; Volume 11, pp. 189–217. [Google Scholar]
- Patel, N.; Patel, S.; Joshi, A. Multiple unit pellet system (MUPS technology) for development of modified release fast disintegrating tablets: A review. J. Pharm. Sci. Innov. 2017, 6, 50–56. [Google Scholar] [CrossRef]
- Lakio, S.; Tajarobi, P.; Wikström, H.; Fransson, M.; Arnehed, J.; Ervasti, T.; Simonaho, S.P.; Ketolainen, J.; Folestad, S.; Abrahmsén-Alami, S. Achieving a robust drug release from extended release tablets using an integrated continuous mixing and direct compression line. Int. J. Pharm. 2016, 511, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-L.; Pérez, M.B.; Cao, C.; Beer, S.D. Switching (bio-) adhesion and friction in liquid by stimulus responsive polymer coatings. Eur. Polym. J. 2021, 147, 110298. [Google Scholar] [CrossRef]
- Joshi, S.; Petereit, H.U. Film coatings for taste masking and moisture protection. Int. J. Pharm. 2013, 457, 395–406. [Google Scholar] [CrossRef]
- Korasa, K.; Vrečer, F. Overview of PAT process analysers applicable in monitoring of film coating unit operations for manufacturing of solid oral dosage forms. Eur. J. Pharm. Sci. 2018, 111, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Ayenew, Z.; Puri, V.; Kumar, L.; Bansal, A.K. Trends in pharmaceutical taste masking technologies: A patent review. Recent Pat. Drug Deliv. Formul. 2009, 3, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Sun, N.; Wu, B.; Yin, C.; Wu, W. Sigmoidal release of indomethacin from pectin matrix tablets: Effect of in situ crosslinking by calcium cations. Int. J. Pharm. 2006, 318, 132–138. [Google Scholar] [CrossRef]
- Smrdel, P.; Cerne, M.; Bogataj, M.; Urleb, U.; Mrhar, A. Enhanced therapeutic effect of LK-423 in treating experimentally induced colitis in rats when administered in colon delivery microcapsules. J. Microencapsul. 2010, 27, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.F. Site-specific drug delivery systems within the gastro-intestinal tract: From the mouth to the colon. Int. J. Pharm. 2010, 395, 44–52. [Google Scholar] [CrossRef]
- Fallingborg, J. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull. 1999, 46, 183–196. [Google Scholar]
- Humberstone, A.J.; Porter, C.J.; Charman, W.N. A physicochemical basis for the effect of food on the absolute oral bioavailability of halofantrine. J. Pharm. Sci. 1996, 85, 525–529. [Google Scholar] [CrossRef]
- Pouton, C.W.; Porter, C.J. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef]
- Piyakulawat, P.; Praphairaksit, N.; Chantarasiri, N.; Muangsin, N. Preparation and evaluation of chitosan/carrageenan beads for controlled release of sodium diclofenac. AAPS PharmSciTech 2007, 8, E97. [Google Scholar] [CrossRef]
- Arthrotec®. Arthrotec SmPC Information. Available online: https://www.medicines.org.uk/emc/medicine/10673 (accessed on 12 November 2023).
- Bettini, R.; Acerbi, D.; Caponetti, G.; Musa, R.; Magi, N.; Colombo, P.; Cocconi, D.; Santi, P.; Catellani, P.L.; Ventura, P. Influence of layer position on in vitro and in vivo release of levodopa methyl ester and carbidopa from three-layer matrix tablets. Eur. J. Pharm. Biopharm. 2002, 53, 227–232. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, D.W.; Kuk, Y.M.; Park, C.W.; Rhee, Y.S.; Oh, T.O.; Weon, K.Y.; Park, E.S. Investigation of an active film coating to prepare new fixed-dose combination tablets for treatment of diabetes. Int. J. Pharm. 2012, 427, 201–208. [Google Scholar] [CrossRef]
- He, W.; Li, Y.; Zhang, R.; Wu, Z.; Yin, L. Gastro-floating bilayer tablets for the sustained release of metformin and immediate release of pioglitazone: Preparation and in vitro/in vivo evaluation. Int. J. Pharm. 2014, 476, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Zannou, E.A.; Li, P.; Tong, W.-Q. Chapter 40-Product Lifecycle Management (LCM). In Developing Solid Oral Dosage Forms; Qiu, Y., Chen, Y., Zhang, G.G.Z., Liu, L., Porter, W.R., Eds.; Academic Press: San Diego, CA, USA, 2009; pp. 911–921. [Google Scholar]
- Abebe, A.; Akseli, I.; Sprockel, O.; Kottala, N.; Cuitiño, A.M. Review of bilayer tablet technology. Int. J. Pharm. 2014, 461, 549–558. [Google Scholar] [CrossRef]
- Sonvico, F.; Conti, C.; Colombo, G.; Buttini, F.; Colombo, P.; Bettini, R.; Barchielli, M.; Leoni, B.; Loprete, L.; Rossi, A. Multi-kinetics and site-specific release of gabapentin and flurbiprofen from oral fixed-dose combination: In vitro release and in vivo food effect. J. Control. Release 2017, 262, 296–304. [Google Scholar] [CrossRef]
- Vaithiyalingam, S.R.; Sayeed, V.A. Critical factors in manufacturing multi-layer tablets--assessing material attributes, in-process controls, manufacturing process and product performance. Int. J. Pharm. 2010, 398, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Chun, M.-H.; Kim, J.Y.; Park, E.-S.; Choi, D.H. Development of a Robust Control Strategy for Fixed-Dose Combination Bilayer Tablets with Integrated Quality by Design, Statistical, and Process Analytical Technology Approach. Pharmaceutics 2021, 13, 1443. [Google Scholar] [CrossRef] [PubMed]
- Vimovo®. Vimovo® 500/20 Professional Information. Available online: https://pi-pill-repository.sahpra.org.za/wp-content/uploads/2022/08/pi_vimovo_21072022.pdf (accessed on 17 November 2023).
- Ambike, A.A.; Mahadik, K.R.; Paradkar, A. Stability study of amorphous valdecoxib. Int. J. Pharm. 2004, 282, 151–162. [Google Scholar] [CrossRef]
- Kolter, K.; Karl, M.; Gryczke, A. Hot-Melt Extrusion with BASF Pharma Polymers, 2nd ed.; BASF: Ludwigshafen, Germany, 2012; pp. 18–30. [Google Scholar]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-Melt Extrusion: From Theory to Application in Pharmaceutical Formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [CrossRef]
- Stanković, M.; Frijlink, H.W.; Hinrichs, W.L. Polymeric formulations for drug release prepared by hot melt extrusion: Application and characterization. Drug Discov. Today 2015, 20, 812–823. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Tiwari, R.V.; Patil, H.; Repka, M.A. Contribution of hot-melt extrusion technology to advance drug delivery in the 21st century. Expert Opin. Drug Deliv. 2016, 13, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Petrović, J.; Ibrić, S.; Betz, G.; Đurić, Z. Optimization of matrix tablets controlled drug release using Elman dynamic neural networks and decision trees. Int. J. Pharm. 2012, 428, 57–67. [Google Scholar] [CrossRef]
- Nish, S.; Mathew, G.; Lincy, J. Matrix Tablets: An Effective Way for Oral Controlled Release Drug Delivery. Iran. J. Pharm. Sci. 2012, 8, 165–170. [Google Scholar]
- Riamet®. Riamet® SmPC. Available online: https://www.medicines.org.uk/emc/product/1628/smpc (accessed on 15 November 2023).
- Genina, N.; Boetker, J.P.; Colombo, S.; Harmankaya, N.; Rantanen, J.; Bohr, A. Anti-tuberculosis drug combination for controlled oral delivery using 3D printed compartmental dosage forms: From drug product design to in vivo testing. J. Control. Release 2017, 268, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Moulton, S.E.; Wallace, G.G. 3-dimensional (3D) fabricated polymer based drug delivery systems. J. Control. Release 2014, 193, 27–34. [Google Scholar] [CrossRef]
- Skowyra, J.; Pietrzak, K.; Alhnan, M.A. Fabrication of extended-release patient-tailored prednisolone tablets via fused deposition modelling (FDM) 3D printing. Eur. J. Pharm. Sci. 2015, 68, 11–17. [Google Scholar] [CrossRef]
- Goole, J.; Amighi, K. 3D printing in pharmaceutics: A new tool for designing customized drug delivery systems. Int. J. Pharm. 2016, 499, 376–394. [Google Scholar] [CrossRef]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Roberts, C.J. Desktop 3D printing of controlled release pharmaceutical bilayer tablets. Int. J. Pharm. 2014, 461, 105–111. [Google Scholar] [CrossRef]
- Katakam, P.; Dey, B.; Assaleh, F.H.; Hwisa, N.T.; Adiki, S.K.; Chandu, B.R.; Mitra, A. Top-Down and Bottom-Up Approaches in 3D Printing Technologies for Drug Delivery Challenges. Crit. Rev. Ther. Drug Carr. Syst. 2015, 32, 61–87. [Google Scholar] [CrossRef]
- Katstra, W.E.; Palazzolo, R.D.; Rowe, C.W.; Giritlioglu, B.; Teung, P.; Cima, M.J. Oral dosage forms fabricated by three dimensional printing. J. Control. Release 2000, 66, 1–9. [Google Scholar] [CrossRef]
- Wang, C.C.; Tejwani Motwani, M.R.; Roach, W.J.; Kay, J.L.; Yoo, J.; Surprenant, H.L.; Monkhouse, D.C.; Pryor, T.J. Development of near zero-order release dosage forms using three-dimensional printing (3-DP) technology. Drug Dev. Ind. Pharm. 2006, 32, 367–376. [Google Scholar] [CrossRef]
- Ventola, C.L. Medical Applications for 3D Printing: Current and Projected Uses. Pharm. Ther. 2014, 39, 704–711. [Google Scholar]
- Ursan, I.D.; Chiu, L.; Pierce, A. Three-dimensional drug printing: A structured review. J. Am. Pharm. Assoc. 2013, 53, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Chai, H.; Wang, X.; Yang, J.; Li, J.; Zhao, Y.; Cai, W.; Tao, T.; Xiang, X. Fused Deposition Modeling (FDM) 3D Printed Tablets for Intragastric Floating Delivery of Domperidone. Sci. Rep. 2017, 7, 2829. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.K.; Lorber, B.; Reitsamer, H.; Khinast, J. 3D printing of oral drugs: A new reality or hype? Expert Opin. Drug Deliv. 2018, 15, 1–4. [Google Scholar] [CrossRef]
- Figueiredo, S.; Fernandes, A.I.; Carvalho, F.G.; Pinto, J.F. Exploring Environmental Settings to Improve the Printability of Paroxetine-Loaded Filaments by Fused Deposition Modelling. Pharmaceutics 2023, 15, 2636. [Google Scholar] [CrossRef]
- PharmaExcipients. Tablet in Capsule Technology–Overview. Available online: https://www.pharmaexcipients.com/news/tablet-capsule-overview/ (accessed on 8 December 2023).
- Bowtle, W.J. Capsule-in-Capsule Technology. Available online: https://www.pharmtech.com/view/capsule-capsule-technology (accessed on 8 December 2023).
- ManufacturingChemist. Delivering Fixed-Dose Combination Therapies with Hard Capsules: Part II. Available online: https://www.manufacturingchemist.com/news/article_page/Delivering_fixed-dose_combination_therapies_with_hard_capsules_part_II/179525 (accessed on 8 December 2023).
- AccuBreak. Developers of Tablets with Drug-Free Break Layers. Available online: https://accubreak.com/tablet-technologies/ (accessed on 10 December 2023).
| Individual Therapeutic Components | Number of Tablets Daily | Fixed-Dose Combination (FDC) | Number of Tablets Daily |
|---|---|---|---|
| Intensive phase: scenario one | |||
| Rifampicin (600 mg) | 1 | Rifampicin + Isoniazid + Pyrazinamide + Ethambutol (150/75/400/275 mg) | 4 |
| Isoniazid (300 mg) | 1 | ||
| Pyrazinamide (500 mg) | 3 | ||
| Ethambutol (400 mg) | 2 | ||
| Total: | 7 | Total: | 4 |
| Intensive phase: scenario two | |||
| Rifampicin (150 mg) | 4 | Rifampicin + Isoniazid + Pyrazinamide + Ethambutol (150/75/400/275 mg) | 4 |
| Isoniazid (100 mg) | 3 | ||
| Pyrazinamide (500 mg) | 3 | ||
| Ethambutol (400 mg) | 2 | ||
| Total: | 12 | Total: | 4 |
| Maintenance phase: scenario one | |||
| Rifampicin (600 mg) | 1 | Rifampicin + Isoniazid (300/150 mg) | 2 |
| Isoniazid (300 mg) | 1 | ||
| Total: | 2 | Total: | 2 |
| Maintenance phase: scenario two | |||
| Rifampicin (150 mg) | 4 | Rifampicin + Isoniazid (300/150 mg) | 2 |
| Isoniazid (100 mg) | 3 | ||
| Total: | 7 | Total: | 2 |
| Brand Name(s) | Active Pharmaceutical Ingredients | Indication |
|---|---|---|
| CV-Pill kit (Torrent Pharmaceuticals) | Metoprolol, ramipril, aspirin, atorvastatin 50/5/75/10 mg | Primary prevention |
| Heart Pill (Excella Pharma) | Ramipril, aspirin, atorvastatin 2.5/100/4; 5/100/40; 10/100/40 mg | Primary prevention |
| Polycap™ (Cadila Pharmaceuticals) | Atenolol, hydrochlorothiazide, ramipril, aspirin, simvastatin 50/12.5/5/100/20 mg | Primary prevention |
| Polypill-E (Alborz Darou Pharmaceuticals) | Enalapril, hydrochlorothiazide, aspirin, atorvastatin 2.5/12.5/81/20 mg | Primary prevention |
| Polypill-V (Alborz Darou Pharmaceuticals) | Hydrochlorothiazide, valsartan, aspirin, atorvastatin 12.5/40/81/20 mg | Primary prevention |
| Polytorva® (USV) | Ramipril, aspirin, atorvastatin 10/75/5 mg | Secondary prevention |
| Ramitorva™ (Zydus Cadila Healthcare) | Ramipril, aspirin, atorvastatin 5/75/10 mg | Secondary prevention |
| Red Heart Pill 1 (Dr Reddy’s Laboratories) | Atenolol, lisinopril, aspirin, simvastatin 50/10/75/40 mg | Secondary prevention |
| Red Heart Pill 2 (Dr Reddy’s Laboratories) | Lisinopril, hydrochlorothiazide, aspirin, simvastatin 10/12.5/75/40 mg | Secondary prevention |
| Starpill (Cipla) | Atenolol, losartan, aspirin, atorvastatin 50/50/75/10 mg | Secondary prevention |
| Trinomia® (Ferrer) | Ramipril, aspirin, atorvastatin 2.5/100/40; 5/100/40; 10/100/40 mg | Secondary prevention |
| ZYCAD-4 kit (Zydus Cadila Healthcare) | Ramipril, aspirin, atorvastatin, metoprolol 5/75/100/50 mg | Secondary prevention |
| Brand Name(s) | Active Pharmaceutical Ingredients | Classification |
|---|---|---|
| Acesyl Co® (Akacia); Ariprel Plus® (Watson Pharma); Coversyl® Plus (Servier); Pearinda Plus® (Pharma Dynamics); Perindopril Co Unicorn®; Prexum Plus® (Biogaran); Vectoryl Plus® (Aspen) | Perindopril, indapamide 4/1.25 mg | ACE inhibitor + diuretic [28] |
| Accumax Co® (Pfizer); Accuretic® (Pfizer); Adco-Quinaretic® | Quinapril, hydrochlorothiazide 10/12.5; 20/12.5; 20/25 mg | ACE inhibitor + diuretic [28] |
| Aldazide (Pfizer) | Spironolactone, isobutyl hydrochlorothiazide 25/2.5 mg | Aldosterone antagonist + diuretic [28] |
| Altoran®-CH (Alembic); Arbozil™-CT (Zuventus); Asar®-CT (Glenmark); Edarbyclor® (Takeda Pharmaceuticals; Valeant); Myotan®-CT (Synokem); Tezihart-CH (Leeford) | Azilsartan, chlorthalidone 40/12.5; 40/25 mg | Angiotensin-receptor blocking + thiazide-like diuretic [28] |
| Amilorectic® (aspen); Moduretic® (MSD); Adco-retic®, Betaretic® (Ranbaxy) | Amiloride, hydrochlorothiazide 5/50; 2.5/25 mg | Potassium sparing agents + diuretic [28] |
| Amlodipine and benazepril (Dr Reddy’s Laboratories); amlodipine and benazepril (Watson); Benidep (Johnlee); benazepril and amlodipine (Systopic Laboratories) |
Benazepril, amlodipine
10/2.5; 10/5; 20/5; 20/10; 40/5; 40/10 mg | ACE inhibitor + calcium-channel blockers [28] |
| Atacand Plus® (AstraZeneca) | Candesartan, hydrochlorothiazide 16/12.5; 32/12.5; 32/25 mg | Angiotensin II antagonist + diuretic [28,62] |
| Atamra CV kit (Amra Remedies) | Atorvastatin, ramipril, clopidogrel 10/5/75 mg | HMG CoA reductase inhibitor, ACE inhibitor, platelet aggregation inhibitor [14] |
| Caduet (Pfizer) | Amlodipine, atorvastatin 5/10; 5/20; 5/40; 5/80; 10/10; 10/20; 10/40; 10/80 mg | Calcium channel blocker + HMG CoA reductase inhibitor [28] |
| Cibadrex® (Novartis) | Benazepril, hydrochlorothiazide 10/12.5 mg | ACE inhibitor + diuretic [28] |
| Coaprovel® (Sanofi-Aventis); Co-Irbewin® (Withrop); Isart Co® (Zydus) | Irbesartan, hydrochlorothiazide 150/12.5; 300/12.5 mg | Angiotensin II antagonist + diuretic [28] |
| Co-Renitec® (MSD), Enap-Co® (Pharma Dynamics), Pharmapress Co® (Aspen) | Enalapril, hydrochlorothiazide 20/12.5 mg | ACE inhibitor + diuretic [28] |
| Cozaar Comp® (MSD); Lohype Plus® (Ranbaxy Betabs); Losacar Co® (Zydus); Ciplazar Co® (Cipla) | Losartan, hydrochlorothiazide 50/12.5 mg | Angiotensin II antagonist + diuretic [28] |
| Co-Pritor® (Ingelheim); Co-Micardis® (Ingelheim) | Telmisartan/hydrochlorothiazide 40/12.5; 80/12.5 mg; 80/25 mg | Angiotensin II antagonist + diuretic [28] |
| Co-Diovan® (Novartis); Co-Tareg® (Novartis); Co-Zomevek® (Novartis) | Valsartan/hydrochlorothiazide 80/12.5; 160/12.5; 160/25 mg; 320/12.5; 320/25 mg | Angiotensin II antagonist + diuretic [28] |
| Co Exforge® (Novartis) | Losartan, amlodipine, hydrochlorothiazide 160/5/212.5/; 160/10/12.5; 160/5/25; 160/10/25 mg | Angiotensin II antagonist + calcium-channel blocker [28] |
| Dyazide® (Litha Pharma); Renezide® (Aspen) | Triamterene, hydrochlorothiazide 50/25 mg | Potassium-sparing agent + diuretic [28] |
| Exforge® (Novartis) | Valsartan, amlodipine 160/5; 320/5; 160/10; 320/10 mg | Angiotensin II antagonist + calcium-channel blocker [28] |
| Entresto™ (Novartis); Vymada® (Novartis); Valsa 50 (Natco); Azmarda® (Cipla); Valcubit® (Elder) | Sacubitril, valsartan 24/26; 49/51; 97/103 mg | Neprilysin inhibitor + angiotensin II antagonist [28] |
| Fosinopril and hydrochlorothiazide (Biogaran®; Cipla; Citron; Glenmark; Ranbaxy; Rising®) | Fosinopril, hydrochlorothiazide 10/12.5; 20/12.5 mg | ACE inhibitor + diuretic [28] |
| Fortzaar® (MSD); Lohype Forte Plus® | Losartan, hydrochlorothiazide 100/25 mg | Angiotensin II antagonist + diuretic [28] |
| Imprida® HCT (Novartis) | Amlodipine, valsartan, hydrochlorothiazide 5/160/12.5; 10/320/25 mg | Calcium channel blocker + angiotensin II antagonist + diuretic [14] |
| Inhibace® Plus (Roche) | Cilazapril, hydrochlorothiazide 5/12.5 mg | ACE inhibitor + diuretic [28] |
| Livper-A (Livealth); Perindopril- amlodipine-Mepha® (Mepha); Perindopril- amlodipine-STADA (Stada); Perindopril- amlodipine teva (Teva) |
Perindopril, amlodipine
3.5/2.5; 7/5; 14/10 mg | ACE inhibitor + calcium-channel blockers [28] |
| Losaar Plus® (Accord); Sartoc Co (Aspen); Zartan Co® (Pharma Dynamics); Hytenza Co® (Watson Pharma); Netrasol® (Specpharm) | Losartan, hydrochlorothiazide 50/12.5; 100/25 mg | Angiotensin II antagonist + diuretic [28] |
| Preterax® (Servier) | Perindopril, indapamide 2/0.625 mg | ACE inhibitor + diuretic [28] |
| Polypill (Cipla) | Amlodipine, losartan, hydrochlorothiazide, simvastatin 2.5/25/12.5/40 mg | Calcium-channel blocker, angiotensin II antagonist, diuretic, HMG CoA reductase inhibitor [25] |
| RIL–AA (East West Pharma) | Ramipril, atorvastatin, aspirin 5/10/75 mg | ACE inhibitor, HMG CoA reductase inhibitor, platelet aggregation inhibitor [64] |
| Servatrin® (Aspen) | Timolol, amiloride, hydrochlorothiazide 10/2.5/25 mg | β-blocker, non-selective, potassium-sparing agents + diuretic [28] |
| Spec-Perindopril Plus® | Perindopril, indapamide 2/0.625; 4/1.25 mg | ACE inhibitor + diuretic [28] |
| Tarka® (Abbott) | Trandolapril, verapamil 2/180; 4/240 mg | ACE inhibitor + calcium-channel blockers [28] |
| Tenoretic® (AstraZeneca); Sandoz Co-tenidone® 100/25, 50/12.5; Tenchlor® (Aspen); Tenoret 50® (AstraZeneca); Tenchlor HS® (Aspen) | Atenolol, chlortalidone 100/25; 50/12.5 mg | β-blockers, selective + diuretic [28] |
| Tri-Plen® (Sanofi-Aventis) | Ramipril, felodipine 2.5/2.5; 5/5 mg | ACE inhibitor + calcium-channel blockers [28] |
| Triplixan® (Servier) | Amlodipine; perindopril, indapamide 5/5/1.25; 10/10/2.5 mg | Calcium channel blocker + ACE inhibitor + diuretic [14] |
| Tritace Plus® (Sanofi-Aventis) | Ramipril, hydrochlorothiazide 2.5/12.5; 5/12.5; 10/25 mg | ACE inhibitor + diuretic [28] |
| Triveram® (Servier) | Perindopril, amlodipine, atorvastatin 10/40/10 mg | ACE inhibitor, calcium-channel blocker, HMG CoA reductase inhibitor [14] |
| Twynsta® (Ingelheim) | Telmisartan, amlodipine 40/5; 80/5; 40/10; 80/10 mg | Angiotensin II antagonist + calcium-channel blocker [28] |
| Uniretic® (Schwarz Pharma’s) | Moexipril, hydrochlorothiazide 7.5/12.5; 15/12.5; 15/25 mg | ACE inhibitor + diuretic [28] |
| Zaneril® (Litha Pharma) | Enalapril, lercanidipine 10/10; 20/10 mg | ACE inhibitor + calcium-channel blockers [28] |
| Zapto-Co® (Aspen) | Captopril, hydrochlorothiazide 50/12.5 mg | ACE inhibitor + diuretic [28] |
| Zestoretic® (AstraZeneca); Auro-Lisinopril Co® (Actor Pharma); Hexal-Lisinopril Co® (Sandoz); Lisinopril Co Unicorn®; Lisoretic® (Pharma Dynamics); Lisinozide® (Novagen); Lisozide (Austell); Diace Co® (Simayla); Zestozide® (Mylan) | Lisinopril, hydrochlorothiazide 10/12.5; 20/12.5; 20/25 mg | ACE inhibitor + diuretic [28] |
| Ziak® (Merck); Bilocor Co® (Pharma dynamic); Bisoprolol Hydrochlorothiazide Zydus® (Zydus) | Bisoprolol, hydrochlorothiazide 2.5/6.25; 5/6.25; 10/6.25 mg | Beta-blockers (selective) + diuretic [28] |
| Brand Name(s) | Active Pharmaceutical Ingredients | Classification |
|---|---|---|
| Macron® (Mylan); Megapen® (Aspen) | Amoxicillin, flucloxacillin 250/250 mg | Beta-lactam sensitive penicillins with extended spectrum |
| Apen® (Mylan) | Ampicillin, cloxacillin 250/250 mg | |
| Augmentin® (Aspen); Adco-amoclav® (AI Pharm); Amoclan® (Watson pharma); AugMaxil® (Aspen); Ranclav® (Ranbaxy); Clamentin® (Mylan); Austell-Co-Amoxiclav®; Bindoclav® (Actor Pharma) | Amoxicillin, clavulanic acid 250/125; 500/125; 875/125 mg | Beta-lactam inhibitors and penicillins |
| Bactrim® (Roche); Adco-Co-Trimoxazole®; Lagatrim® (Akacia); Nucotrim® (GulfDrug); Cozole® (Ranbaxy); Purbac® (Aspen) | Trimethoprim, sulfamethoxazole 80/400 mg | Trimethoprim and sulphonamides |
| Brand Name(s) | Active Pharmaceutical Ingredients | Classification |
|---|---|---|
| Coartem® (Novartis), Artefan® (Ajanta), Lumet (Cipla), Lumerax (IPCA Laboratories Ltd.), Combiart® (Strides Arcolab Limited), Lumiter (Macleods), Komefan (Mylan), Riamet® (Novartis) | Artemether, lumefantrine 20/120 mg | Artemisinin + fluorene [28,68] |
| Pyramax® (co-developed by MMV and Shin Poong Pharmaceutical) | Artesunate, pyronaridine 60/180 mg | Artemisinin + benzonaphthyridine derivative [69] |
| MEFLIAM (Cipla); Wellcigo Plus (Wellona Pharma); Falcigo Plus (Zydus Cadila) | Artesunate, mefloquine 25/50; 100/200 mg | Artemisinin + analogue of quinine [68] |
| Coarsucam™ (Sanofi-Aventis); Artesunate/Amodiaquine Winthrop® tablet | Artesunate, amodiaquine 25/67.5; 50/135; 100/270 mg | Artemisinin + quinoline [68] |
| Artecospe® (Guilin Pharmaceutical) | Artesunate, sulfadoxine, pyrimethamine 50/500/25 mg | Artemisinin + sulfonamide + folic acid antagonist |
| Fansidar® (Akacia; Roche; Ascendis Pharma) | Sulfadoxine, pyrimethamine 250/12.5; 500/25 mg | Sulfonamide + folic acid antagonist [28,68] |
| Eurartesim (Alfasigma), Artekin (Holleykin), Duocotexin (Holley Pharm) | Dihydroartemisinin, piperaquine 10/80 mg | Artemisinin + aminoquinoline [68] |
| Malarone® and Malanil™ (GlaxoSmithKline) | Atovaquone, proguanil hydrochloride 250/100 mg | Naphthoquinones + biguanide derivative [28] |
| SynriamTM (Ranbaxy) | Arterolane maleate, piperaquine phosphate 150/750 mg | Adamantanes + aminoquinoline [70] |
| Brand Name(s) | Active Pharmaceutical Ingredients | Indication |
|---|---|---|
| Isonarif TM (Versa Pharma); Rifamate® (Sanofi); Rifinah (Sanofi-Aventis); Riwell-IS (Wellona pharma) | Rifampicin, isoniazid 150/75; 300/150 mg | First-line drugs in combination for tuberculosis treatment [5,28] |
| Rimactazid (Sandoz) | Rifampicin, isoniazid 150/75; 300/150; 60/60 mg | |
| Combunex 800 (Lupin); Ethox-IN (Talent healthcare) | Ethambutol, isoniazid 800/300 mg | |
| Rifater (Sanofi-Aventis); RismidCare (AdvaCare) | Rifampin, isoniazid, pyrazinamide 120/50/300 mg | |
| Onecure (TGP); Rifafour e-275 (Sanofi-Aventis) | Rifampicin, isoniazid, pyrazinamide, ethambutol 150/75/400/275 mg |
| Brand Name(s) and Companies | Minimum Body Weight, Weight range, or age | Active Pharmaceutical Ingredients Classification | ||||
|---|---|---|---|---|---|---|
| NRTIs/NtRTIs | NNRTIs | INSTI | PI | PK Enhancer | ||
| Cimduo® (Mylan); Temixys™ (Janssen) | ≥35 kg | Lamivudine (300 mg) Tenofovir disoproxil (300 mg) | ||||
| Combivir® (GlaxoSmithKline, ViiV); Combozil (HeteroDrugs SA); Duovir (Cipla); Adco- lamivudine and zidovudine; Lamzid (Aspen); Loziv (Novagen Pharma) | 30 kg | Lamivudine (150 mg) Zidovudine (300 mg) | ||||
| Descovy® (Gilead) | 14 to < 25 kg | Emtricitabine (120 mg) Tenofovir alafenamide (15 mg) | ||||
| 25–35 kg | Emtricitabine (200 mg) Tenofovir alafenamide (25 mg) | |||||
| Epzicom® (US, ViiV), Kivexa® (GSK, ViiV Healthcare); Dumiva (Mylan) | 25 kg | Abacavir (600 mg) Lamivudine (300 mg) | ||||
| Truvada® (Gilead); Adco-Emtevir (Adcock Ingrams); Tencitab (Aspen); Didivir (Cipla); Tyricten (Aurobindo); Tenemine (Mylan) | 17 to <22 kg | Emtricitabine (100 mg) Tenofovir disoproxil (150 mg) | ||||
| 22 to <28 kg | Emtricitabine (133 mg) Tenofovir disoproxil (200 mg) | |||||
| 28 to <35 kg | Emtricitabine (167 mg) Tenofovir disoproxil (250 mg) | |||||
| 35 kg | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | |||||
| Atripla® (MSD); Atroiza (Mylan); Citenvir (Novagen); Odimune (Cipla); Tribuss™ (Aspen) | 40 kg | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | Efavirenz (600 mg) | |||
| Complera® (Aspen), Eviplera® (Gilead) | 35 kg and aged ≥12 years | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | Rilpivirine (25 mg) | |||
| Delstrigo® (Merck & Co.) | 35 kg | Lamivudine (300 mg) Tenofovir disoproxil (300 mg) | Doravirine (100 mg) | |||
| Odefsey® (Aspen) | 35 kg and aged ≥12 years | Lamivudine (300 mg) Tenofovir disoproxil (300 mg) | Rilpivirine (25 mg) | |||
| Symfi® (Mylan); Tenarenz (Aspen) | 40 kg | Lamivudine (300 mg) Tenofovir disoproxil (300 mg) | Efavirenz (600 mg) | |||
| Biktarvy® (Aspen) | 14 to < 25 kg | Emtricitabine (120 mg) Tenofovir alafenamide (15 mg) | Bictegravir (30 mg) | |||
| 25 kg | Emtricitabine (200 mg) Tenofovir alafenamide (25 mg) | Bictegravir (50 mg) | ||||
| Dovato (GlaxoSmithKline; ViiV Healthcare) | Minimum weight for individual components | Lamivudine (300 mg) | Dolutegravir (50 mg) | |||
| Triumeq® (GlaxoSmithKline) | 25 kg | Abacavir (600 mg) Lamivudine (300 mg) | Dolutegravir (50 mg) | |||
| Triumeq® PD (GlaxoSmithKline) | 10 to <25 kg | Abacavir (60 mg) Lamivudine (300 mg) | Dolutegravir (50 mg) | |||
| Genvoya® (Aspen) | 25 kg | Emtricitabine (200 mg) Tenofovir alafenamide (10 mg) | Elvitegravir (150 mg) | Cobicistat (150 mg) | ||
| Genvoya® (Aspen) | 35 kg and SMR 4 or 5 | Emtricitabine (200 mg) Tenofovir disoproxil (300 mg) | Elvitegravir (150 mg) | Cobicistat (150 mg) | ||
| Evotaz® (Bristol-Myers Squibb) | 35 kg | Atazanavir (300 mg) | Cobicistat (150 mg) | |||
| Prezcobix® (Janssen) | >40 kg | Darunavir (800 mg) | Cobicistat (150 mg) | |||
| Kaletra® (AbbVie) | >40 kg | Lopinavir (100 mg, 200 mg tablets) | Ritonavir (25 mg, 50 mg tablets) | |||
| Brand Name and Company | Combination Drugs | Classification |
|---|---|---|
| Acarjohn-M (Johnlee Pharmaceuticals) | Acarbose, metformin 25/500 mg | Alpha-glucosidase inhibitors + biguanide [102] |
| Avandamet® (GSK) | Rosiglitazone, metformin 2/500; 4/500; 2/1000; 4/1000 mg | Thiazolidinediones + biguanide [101,102] |
| Janumet® (MSD) | Sitagliptin, metformin 50/500; 50/850; 50/1000 mg | Dipeptidyl peptidase 4 inhibitor + biguanide [28,101,102] |
| Amaryl® (Sanofi-Aventis) | Glimepiride, metformin 1/500; 2/500 mg | Sulfonylureas + biguanide [28,101,102] |
| Glucovance® (Merck) | Glibenclamide, metformin 1.25/500; 2.5/500; 5/500 mg | |
| MetaglipTM (Bristol-Myers Squibb) | Glipizide, metformin 2.5/250; 2.5/500 mg | |
| Galvus Met® (Novartis) | Vildagliptin, metformin 50/1000; 50/850 mg | Dipeptidyl peptidase 4 inhibitor + biguanide [28,101] |
| PrandiMet® (Sciele and Novo Nordisk) | Repaglinide, metformin 1/500; 2/500 mg | Meglitinides + biguanide |
| Avandaryl® (GlaxoSmithKline) | Rosiglitazone, glimepiride 4/1; 4/2; 4/4; 8/2; 8/4 mg | Thiazolidinediones + sulfonylureas [102] |
| Duetact® (JPI, Takeda Pharmaceuticals) | Pioglitazone, glimepiride 30/2; 30/4 mg | |
| Jentadueto® (Boehringer Ingelheim) | Linagliptin, metformin 2.5/850; 2.5/1000 mg | Dipeptidyl peptidase 4 inhibitors + biguanide [101] |
| Kazano (Takeda Pharmaceuticals) | Aloglipine, metformin 12.5/500; 12.5/1000 mg | |
| Komboglyze® (AstraZeneca & Bristol-Myers Squibb) | Saxagliptin, metformin 2.5/500; 2.5/850; 2.5/1000 mg | |
| InvokametTM (Janssen Pharmaceuticals) | Canagliflozin, metformin 50/500; 50/850; 50/1000; 150/500; 150/850; 150/1000 mg | Sodium-glucose co-transporter 2 inhibitors + biguanide [101,102] |
| Xigduo® (AstraZeneca) | Dapagliflozin, metformin 5/850; 5/1000 mg | |
| Synjardy® (Boehringer Ingelheim) | Empagliflozin, metformin 5/500; 5/850; 5/1000; 12.5/500; 12.5/850; 12.5/1000 mg | |
| Segluromet® (Pfizer) | Ertugliflozin, metformin 2.5/850; 2.5/1000; 7.5/850; 7.5/1000 mg | |
| Glyxambi® (Boehringer Ingelheim) | Empagliflozin, linagliptin 10/5; 25/5 mg | Dipeptidyl peptidase 4 inhibitors + sodium- glucose co-transporter 2 inhibitors [101,102] |
| Qtern® (AstraZeneca) | Saxagliptin, dapagliflozin 5/10 mg | |
| Steglujan® (Merck) | Sitagliptin, ertugliflozin 5/100; 15/100 mg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkins, C.A.; Hamman, H.; Hamman, J.H.; Steenekamp, J.H.
Fixed-Dose Combination Formulations in Solid Oral Drug
Wilkins CA, Hamman H, Hamman JH, Steenekamp JH.
Fixed-Dose Combination Formulations in Solid Oral Drug
Wilkins, Christi A., Hannlie Hamman, Josias H. Hamman, and Jan H. Steenekamp.
2024. "Fixed-Dose Combination Formulations in Solid Oral Drug