


Development of Mechanistic In Vitro–In Vivo Extrapolation to Support Bioequivalence Assessment of Long-Acting Injectables

 , , and
, , and
Abstract
:1. Introduction
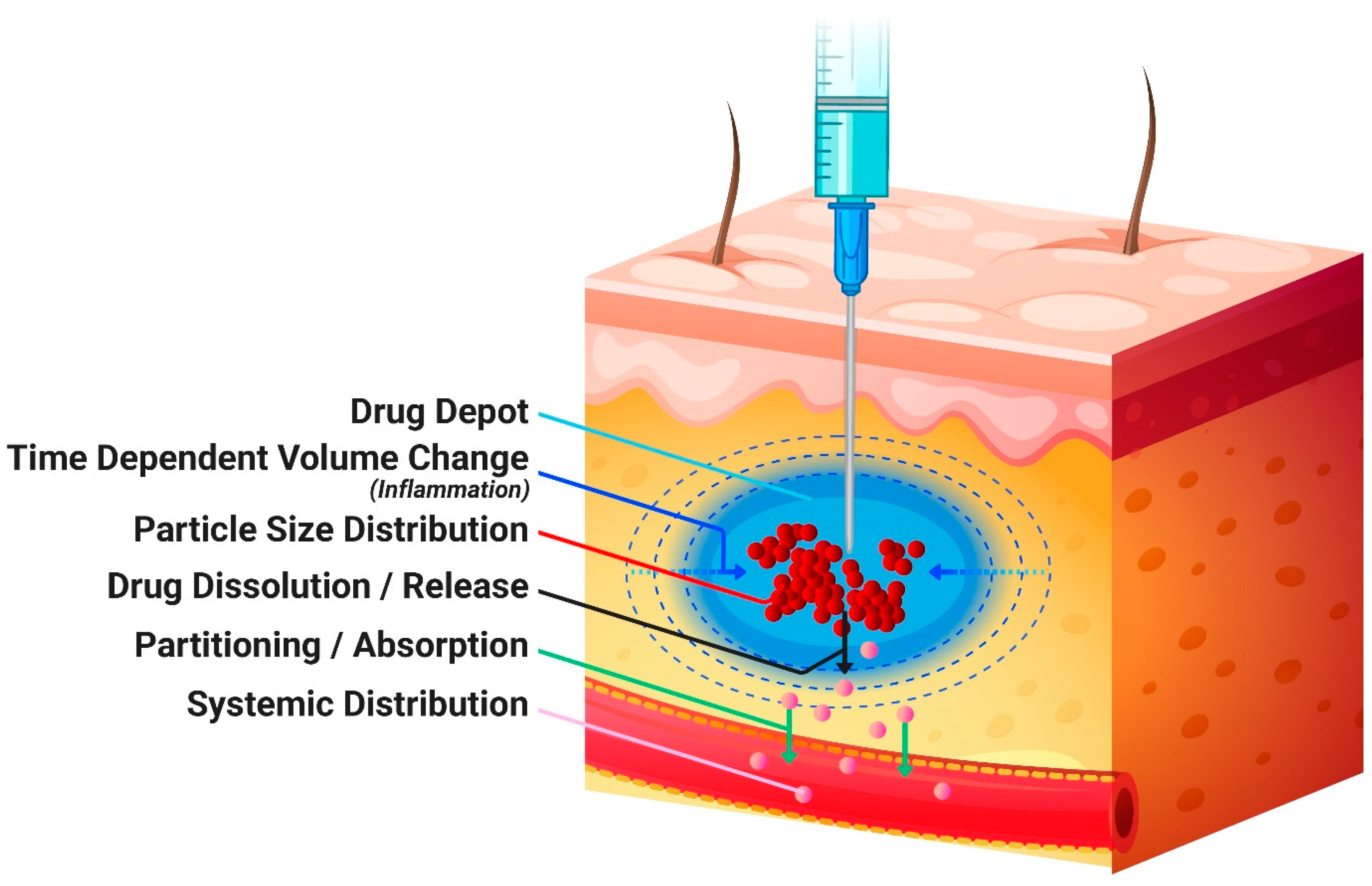
2. Materials and Methods
2.1. In Vitro Characterization and Pre-Clinical In Vivo Studies
2.2. Physiologically-Based Pharmacokinetic Model
2.2.1. Preclinical PBPK Model
2.2.2. Human PBPK Model
2.3. SC Model
2.3.1. Preclinical SC Model
2.3.2. Human SC Model
2.4. In Vitro and In Vivo PSD
2.5. Acceptance Criteria for Model Validation
3. Results
3.1. Preclinical PBPK Model
3.2. Preclinical SC Model and In Vitro–In Vivo Extrapolation
3.3. SC Model in Humans
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Brien, M.N.; Jiang, W.; Wang, Y.; Loffredo, D.M. Challenges and Opportunities in the Development of Complex Generic Long-Acting Injectable Drug Products. J. Control. Release 2021, 336, 144–158. [Google Scholar] [CrossRef]
- Bao, Q.; Zou, Y.; Wang, Y.; Choi, S.; Burgess, D.J. Impact of Formulation Parameters on In Vitro Release from Long-Acting Injectable Suspensions. AAPS J. 2021, 23, 42. [Google Scholar] [CrossRef] [PubMed]
- Alam, K. Mechanistic Modeling of Complex Injectables: Recommendations to Navigate Regulatory Challenges. Available online: https://www.fda.gov/media/166586/download (accessed on 7 March 2024).
- Sharan, S.; Fang, L.; Lukacova, V.; Chen, X.; Hooker, A.C.; Karlsson, M.O. Model-Informed Drug Development for Long-Acting Injectable Products: Summary of American College of Clinical Pharmacology Symposium. Clin. Pharmacol. Drug Dev. 2021, 10, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Suarez-Sharp, S.; Pepin, X.J.H.; Flanagan, T.; Zhao, Y.; Kotzagiorgis, E.; Parrott, N.; Sharan, S.; Tistaert, C.; Heimbach, T.; et al. Applications of Physiologically Based Biopharmaceutics Modeling (PBBM) to Support Drug Product Quality: A Workshop Summary Report. J. Pharm. Sci. 2021, 110, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Siemons, M.; Schroyen, B.; Darville, N.; Goyal, N. Role of Modeling and Simulation in Preclinical and Clinical Long-Acting Injectable Drug Development. AAPS J. 2023, 25, 99. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Hageman, M.J. Development of an In Vitro System To Emulate an In Vivo Subcutaneous Environment: Small Molecule Drug Assessment. Mol. Pharm. 2022, 19, 4017–4025. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Hageman, M.J. Development of Drug Release Model for Suspensions in ESCAR (Emulator of SubCutaneous Absorption and Release). AAPS J. 2023, 25, 29. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Wang, X.; Zou, Y.; Wang, Y.; Burgess, D.J. In Vitro Release Testing Method Development for Long-Acting Injectable Suspensions. Int. J. Pharm. 2022, 622, 121840. [Google Scholar] [CrossRef]
- Bao, Q.; Wang, X.; Wan, B.; Zou, Y.; Wang, Y.; Burgess, D.J. Development of in Vitro-in Vivo Correlations for Long-Acting Injectable Suspensions. Int. J. Pharm. 2023, 634, 122642. [Google Scholar] [CrossRef]
- Package Insert—NDA 21-584, Depo-SubQ Provera 104. Revised: 12/2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021583s033s034lbl.pdf (accessed on 11 April 2024).
- Kozutsumi, D.; Kawashima, A.; Sugimoto, T.; Kotohda, Y.; Fujimori, S.; Takami, M.; Kohno, T.; Oikawa, T.; Sugino, E.; Choshi, T.; et al. Pharmacokinetics of 9α-Fluoromedroxyprogesterone Acetate in Rats: Comparison with Medroxyprogesterone Acetate. Biopharm. Drug Dispos. 1999, 20, 277–284. [Google Scholar] [CrossRef]
- Provera Monograph. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/011839s071lbl.pdf (accessed on 11 April 2024).
- Bick, A.J.; Louw-du Toit, R.; Skosana, S.B.; Africander, D.; Hapgood, J.P. Pharmacokinetics, Metabolism and Serum Concentrations of Progestins Used in Contraception. Pharmacol. Ther. 2021, 222, 107789. [Google Scholar] [CrossRef] [PubMed]
- Stalker, D.; Welshman, I.; Pollock, S. Bioavailability of Medroxyprogesterone Acetate from Three Oral Dosage Formulations. Clin. Ther. 1992, 14, 544–552. [Google Scholar]
- Hardman, M.J.; Hull, D. Blood Flow and Fatty Acid Release by Cervical Adipose Tissue of Rabbits. J. Physiol. 1973, 235, 1–8. [Google Scholar] [CrossRef] [PubMed]
- GastroPlus® Manual; Version 9.8.3; Simulations Plus Inc.: Lancaster, CA, USA, 2022.
- Peloso, C.; Trichet, A.P.; Descotes, J.; Richard, J.; Roberge, C.; Lopez-Noriega, A. Evaluation of Loco-Regional Skin Toxicity Induced by an in Situ Forming Depot after a Single Subcutaneous Injection at Different Volumes and Flow Rates in Göttingen Minipigs. Int. J. Mol. Sci. 2021, 22, 9250. [Google Scholar] [CrossRef] [PubMed]
- Solorio, L.; Babin, B.M.; Patel, R.B.; Mach, J.; Azar, N.; Exner, A.A. Noninvasive Characterization of in Situ Forming Implants Using Diagnostic Ultrasound. J. Control. Release 2010, 143, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Jockel, J.P.L.; Roebrock, P.; Shergold, O.A. Insulin Depot Formation in Subcutaneous Tissue. J. Diabetes Sci. Technol. 2013, 7, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Jucker, B.M.; Fuchs, E.J.; Lee, S.; Damian, V.; Galette, P.; Janiczek, R.; Macura, K.J.; Jacobs, M.A.; Weld, E.D.; Solaiyappan, M.; et al. Multiparametric Magnetic Resonance Imaging to Characterize Cabotegravir Long-Acting Formulation Depot Kinetics in Healthy Adult Volunteers. Br. J. Clin. Pharmacol. 2022, 88, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Jucker, B.M.; Alsaid, H.; Rambo, M.; Lenhard, S.C.; Hoang, B.; Xie, F.; Groseclose, M.R.; Castellino, S.; Damian, V.; Bowers, G.; et al. Multimodal Imaging Approach to Examine Biodistribution Kinetics of Cabotegravir (GSK1265744) Long-Acting Parenteral Formulation in Rat. J. Control. Release 2017, 268, 102–112. [Google Scholar] [CrossRef]
- Port, R.E.; Schuster, C.; Port, C.R.; Bachert, P. Simultaneous Sustained Release of Fludarabine Monophosphate and Gd-DTPA from an Interstitial Liposome Depot in Rats: Potential for Indirect Monitoring of Drug Release by Magnetic Resonance Imaging. Cancer Chemother. Pharmacol. 2006, 58, 607–617. [Google Scholar] [CrossRef]
- Lu, A.T.K.; Frisella, M.E.; Johnson, K.C. Dissolution Modeling: Factors Affecting the Dissolution Rates of Polydisperse Powders. Pharm. Res. 1993, 10, 1308–1314. [Google Scholar] [CrossRef]
- Smith, W.C.; Bae, J.; Zhang, Y.; Qin, B.; Wang, Y.; Kozak, D.; Ashraf, M.; Xu, X. Impact of Particle Flocculation on the Dissolution and Bioavailability of Injectable Suspensions. Int. J. Pharm. 2021, 604, 120767. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.A.; Reddy, M.B.; Heikkinen, A.T.; Lukacova, V.; Parrott, N. Physiologically Based Pharmacokinetic Modelling for First-In-Human Predictions: An Updated Model Building Strategy Illustrated with Challenging Industry Case Studies. Clin. Pharmacokinet. 2019, 58, 727–746. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, H.M.; Mrsny, R.J. Improving the Outcomes of Biopharmaceutical Delivery via the Subcutaneous Route by Understanding the Chemical, Physical and Physiological Properties of the Subcutaneous Injection Site. J. Control. Release 2014, 182, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Berkland, C.; Hageman, M.J. Simulating Particle Movement inside Subcutaneous Injection Site Simulator (SCISSOR) Using Monte-Carlo Method. Int. J. Pharm. 2021, 605, 120824. [Google Scholar] [CrossRef] [PubMed]
- Paquette, S.M.; Dawit, H.; Hickey, M.B.; Merisko-Liversidge, E.; Almarsson, Ö.; Deaver, D.R. Long-Acting Atypical Antipsychotics: Characterization of the Local Tissue Response. Pharm. Res. 2014, 31, 2065–2077. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.J.; Jeong, M.Y.; Jeong, H.T.; Kim, M.S.; Park, H.J.; Kim, D.Y.; Lee, H.C.; Song, W.H.; Kim, C.H.; Lee, C.H.; et al. Effect of Particle Size on in Vivo Performances of Long-Acting Injectable Drug Suspension. J. Control. Release 2022, 341, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Darville, N.; van Heerden, M.; Vynckier, A.; de Meulder, M.; Sterkens, P.; Annaert, P.; van den Mooter, G. Intramuscular Administration of Paliperidone Palmitate Extended-Release Injectable Microsuspension Induces a Subclinical Inflammatory Reaction Modulating the Pharmacokinetics in Rats. J. Pharm. Sci. 2014, 103, 2072–2087. [Google Scholar] [CrossRef]
- Darville, N.; van Heerden, M.; Mariën, D.; de Meulder, M.; Rossenu, S.; Vermeulen, A.; Vynckier, A.; de Jonghe, S.; Sterkens, P.; Annaert, P.; et al. The Effect of Macrophage and Angiogenesis Inhibition on the Drug Release and Absorption from an Intramuscular Sustained-Release Paliperidone Palmitate Suspension. J. Control. Release 2016, 230, 95–108. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Input Parameter | Value | Unit | Source |
|---|---|---|---|
| MPA physicochemical properties | |||
| Molecular weight | 386.53 | g/mol | ADMET Predictor a |
| Solubility @ pH 7.4 | 0.27 | μg/mL | [2] |
| Suspension solubility | 0.01 | mg/mL | [2] |
| pKa | N/A (Neutral compound) | ||
| LogP | 3.82 | [12] | |
| Mean precipitation time | 900 | s | GastroPlus® default |
| Diffusion coefficient | 0.62 | cm2/s × 10−5 | ADMET Predictor |
| Human jejunal permeability | 7.307 × 10−4 | cm/s | ADMET Predictor |
| Drug particle density | 1.2 | g/mL | GastroPlus® default |
| Blood/plasma concentration ratio | 0.83 | ADMET Predictor | |
| Fraction unbound in plasma | 12 | % | [13,14] |
| Kp prediction method | Lukacova | Calculated default GastroPlus® method b | |
| Clearance | |||
| Clearance mechanism | Metabolic (ECCS) | [13] | |
| Kidney CL (Rabbit/Human) | 0.056/0.842 | L/h | Calculated as Fup*GFR |
| Systemic liver CL (Rabbit/Human) | 8.78/30.6 | L/h | Fitted to rabbit intravenous PK data [10]/informed by oral data [15] |
| Subcutaneous parameters | |||
| Injection volume c | 0.64 | mL | |
| Effective depot volume (Rabbit/Human) | 3/4.74 | mL | Fitted |
| Fraction unbound in tissue (Rabbit/Human) | 0.1/0.2 | % | Calculated default GastroPlus® method c |
| Blood flow rate (Rabbit/Human) | 16/3.77 | mL/min/100 g SubQ | [16]/GastroPlus® default |
| Inflammation scaling factor (Rabbit/Human) | 0.536/0.03 | Fitted | |
| Inflammation lag | 0 | day | Fitted |
| Inflammation A | 5.583 | Fitted | |
| Inflammation B | 0.409 | Fitted | |
| Diffusion layer thickness (Rabbit/Human) | 80/150 | μm | Fitted |
| Formulation | In Vitro PSD [10] | In Vivo PSD | |||
|---|---|---|---|---|---|
| Mean # Radius (μm) | SD # (μm) | Mean # Radius (μm) | SD # (μm) | Rmin (μm) | |
| RLD | 9.06 | 3.81 | 16.30 | 18.34 | 4.53 |
| F1 | 6.67 | 3.14 | 12.00 | 15.06 | 3.33 |
| F2 | 10.47 | 6.20 | 18.84 | 29.78 | 5.23 |
| F3 | 6.48 | 3.06 | 11.67 | 14.67 | 3.24 |
| F4 * | 9.39 | 3.86 | 16.92 | 18.51 | 4.69 |
| RLD | F1 | F2 | F3 | F4 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Obs. | Sim. | FE | Obs. | Sim. | FE | Obs. | Sim. | FE | Obs. | Sim. | FE | Obs. | Sim. | FE | |
| Scaled diffusion layer thickness and PSD | |||||||||||||||
| Cmax | 8.9 | 6.69 | 0.75 | 9.96 | 7.53 | 0.76 | 8.82 | 5.84 | 0.66 | 7.46 | 7.61 | 1.02 | 6.75 | 5.07 | 0.75 |
| AUC0-t | 9229 | 9053 | 0.98 | 8435 | 9478 | 1.12 | 7659 | 7556 | 0.99 | 8714 | 9527 | 1.09 | 7779 | 8088 | 1.04 |
| Scaled diffusion layer thickness and PSD & Inflammation | |||||||||||||||
| Cmax | 8.9 | 9.28 | 1.04 | 9.96 | 10.98 | 1.1 | 8.82 | 7.72 | 0.88 | 7.46 | 11.13 | 1.49 | 6.75 | 9.107 | 1.35 |
| AUC0-t | 9229 | 8730 | 0.95 | 8435 | 9193 | 1.09 | 7659 | 7273 | 0.95 | 8714 | 9245 | 1.06 | 7779 | 8343 | 1.07 |
| Observed [11] | Simulated | FE | ||
|---|---|---|---|---|
| Same as rabbit | Cmax | 1.276 | 2.289 | 1.79 |
| AUC0-t | 2005.5 | 2680 | 1.34 | |
| Updated model | Cmax | 1.276 | 1.347 | 1.06 |
| AUC0-t | 2005.5 | 2360.8 | 1.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaral Silva, D.; Le Merdy, M.; Alam, K.D.; Wang, Y.; Bao, Q.; Malavia, N.; Burgess, D.; Lukacova, V. Development of Mechanistic In Vitro–In Vivo Extrapolation to Support Bioequivalence Assessment of Long-Acting Injectables. Pharmaceutics 2024, 16, 552. https://doi.org/10.3390/pharmaceutics16040552
Amaral Silva D, Le Merdy M, Alam KD, Wang Y, Bao Q, Malavia N, Burgess D, Lukacova V. Development of Mechanistic In Vitro–In Vivo Extrapolation to Support Bioequivalence Assessment of Long-Acting Injectables. Pharmaceutics. 2024; 16(4):552. https://doi.org/10.3390/pharmaceutics16040552
Chicago/Turabian StyleAmaral Silva, Daniela, Maxime Le Merdy, Khondoker Dedarul Alam, Yan Wang, Quanying Bao, Nilesh Malavia, Diane Burgess, and Viera Lukacova. 2024. "Development of Mechanistic In Vitro–In Vivo Extrapolation to Support Bioequivalence Assessment of Long-Acting Injectables" Pharmaceutics 16, no. 4: 552. https://doi.org/10.3390/pharmaceutics16040552