
Combination of miR159 Mimics and Irinotecan Utilizing Lipid Nanoparticles for Enhanced Treatment of Colorectal Cancer
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Animals
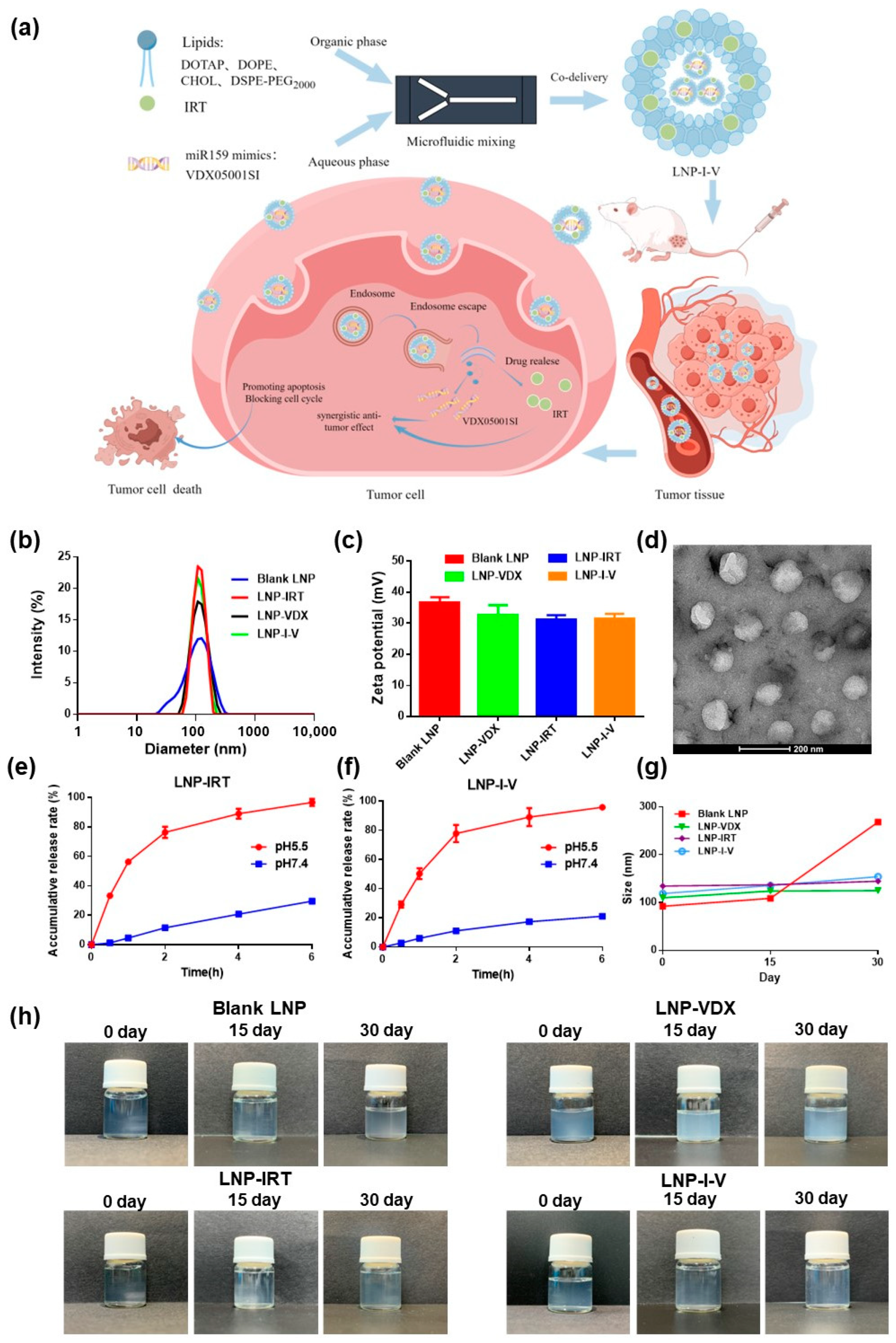
2.4. Preparation and Characterization of LNP-I-V
2.5. In Vitro Release
2.6. Stability of LNP-I-V
2.7. Cell Uptake
2.8. Cytotoxicity
2.9. Apoptosis and Cell Cycle Analysis
2.10. Biodistribution
2.11. In Vivo Anti-Tumor Effect and Immune Analysis
2.12. Biocompatibility
2.13. Statistical Analysis
3. Results
3.1. Preparation and Characterization of LNP-I-V
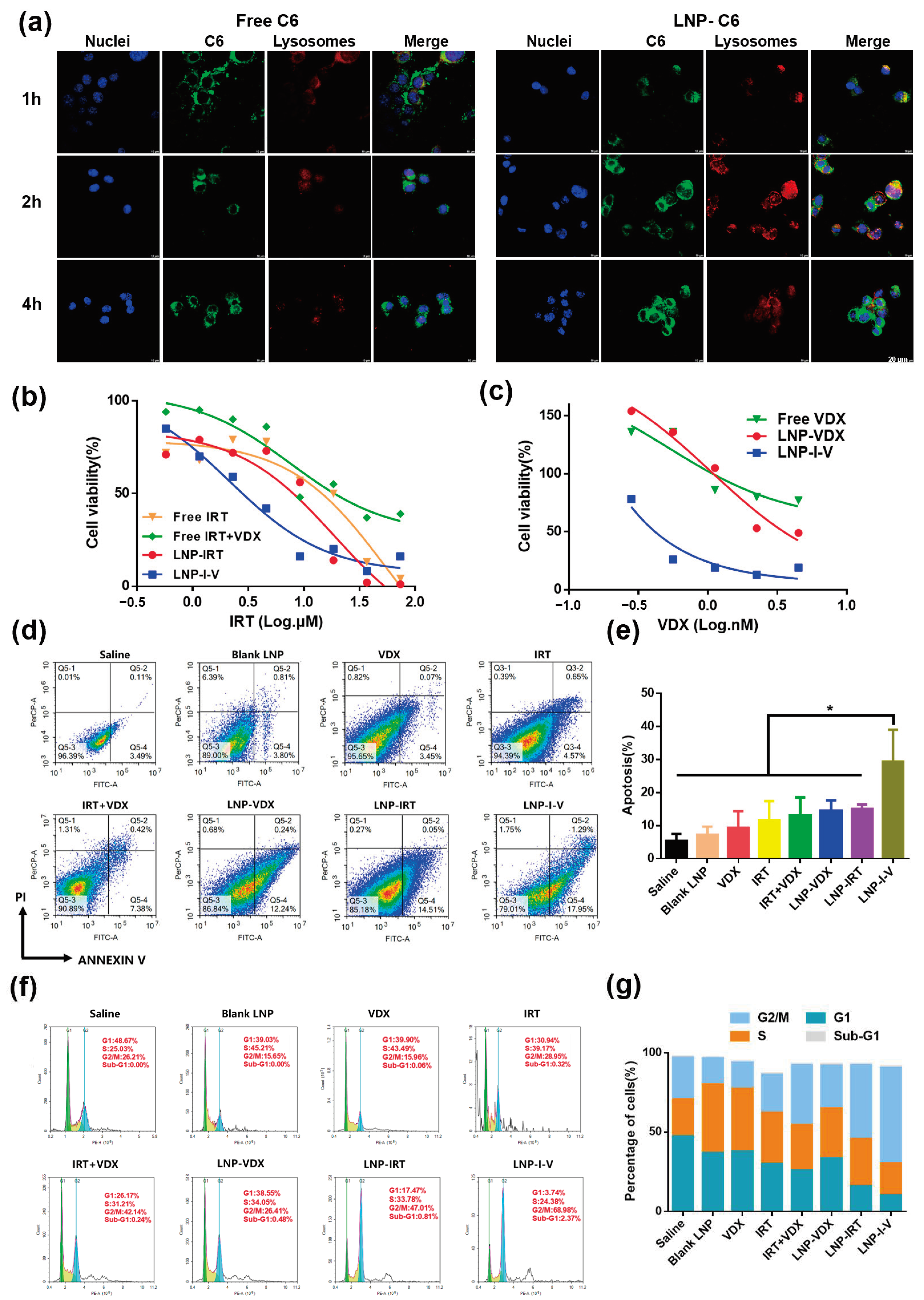
3.2. Cell Uptake
3.3. Cytotoxicity
3.4. Apoptosis and Cell Cycle Analysis
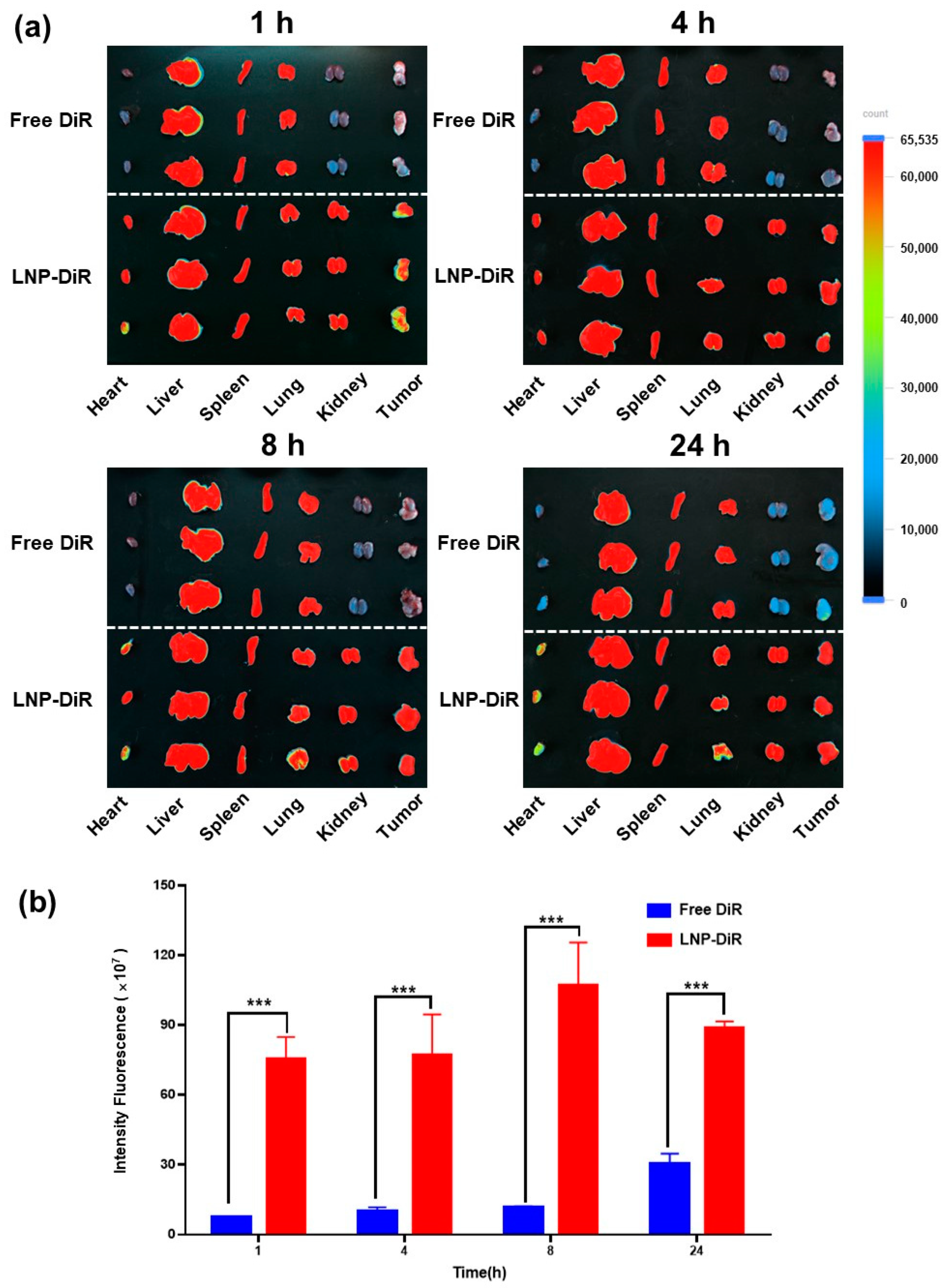
3.5. Biodistribution
3.6. In Vivo Anti-Tumor Efficacy
3.7. Anti-Tumor Immunity
3.8. Biocompatibility
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Liu, N.; Yang, F.; Hu, N.; Wang, M.; Cui, M.; Bruns, N.; Guan, X. Bioengineered protein nanocage by small heat shock proteins delivering mTERT siRNA for enhanced colorectal cancer suppression. ACS Appl. Bio Mater. 2022, 5, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.Q.; Wang, J.; Tan, W.F.; Huang, T.M. Berberine inhibits colorectal tumor growth by suppressing SHH secretion. Acta Pharmacol. Sin. 2021, 42, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Peng, Y.; Ji, F.; Chen, H.; Kang, W.; Chan, L.S.; Gou, H.; Lin, Y.; Huang, P.; Chen, D.; et al. Targeting of SLC25A22 boosts the immunotherapeutic response in KRAS-mutant colorectal cancer. Nat. Commun. 2022, 14, 4677. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, P.; Zhou, M.; Yin, L.; Wang, M.; Liu, T.; Jiang, X.; Gao, H. Small-molecule drugs of colorectal cancer: Current status and future directions. BBA Mol. Basis Dis. 2024, 1870, 166880. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Lee, B.Y.; Kim, K.P. Caffeine enhances chemosensitivity to irinotecan in the treatment of colorectal cancer. Phytomedicine 2023, 121, 155120. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Solanki, R.; Jangid, A.K.; Jain, P.; Pranjali, P.; Patel, S.; Guleria, A.; Pooja, D.; Kulhari, H. Manganese nanocarrier for matrix metalloproteinase 9 responsive delivery of irinotecan for colon cancer treatment. J. Ind. Eng. Chem. 2023, 128, 258–267. [Google Scholar] [CrossRef]
- Ezati, N.; Abdouss, M.; Rouhani, M.; Kerr, P.G.; Kowsari, E. Novel serotonin decorated molecularly imprinted polymer nanoparticles based on biodegradable materials; a potential self-targeted delivery system for irinotecan. React. Funct. Polym. 2022, 181, 105437. [Google Scholar] [CrossRef]
- Yan, W.; Li, Y.; Zou, Y.; Zhu, R.; Wu, T.; Yuan, W.; Lang, T.; Li, Y.; Yin, Q. Co-delivering irinotecan and imiquimod by pH-responsive micelle amplifies anti-tumor immunity against colorectal cancer. Int. J. Pharm. 2023, 648, 123583. [Google Scholar] [CrossRef] [PubMed]
- Pai, F.T.; Lin, W.J. Synergistic cytotoxicity of irinotecan combined with polysaccharide-based nanoparticles for colorectal carcinoma. Biomater. Adv. 2023, 153, 213577. [Google Scholar] [CrossRef]
- Moumné, L.; Marie, A.C.; Crouvezier, N. Oligonucleotide therapeutics: From discovery and development to patentability. Pharmaceutics 2022, 14, 260. [Google Scholar] [CrossRef]
- Thakur, S.; Sinhari, A.; Jain, P.; Jadhav, H.R. A perspective on oligonucleotide therapy: Approaches to patient customization. Front. Pharmcol 2022, 13, 1006304. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Kawamura, A.; Torisu, Y.; Koido, S.; Yahagi, N.; Saruta, M. The clinical potential of oligonucleotide therapeutics against pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 3331. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, J.; Wu, Y.; Dong, Y.; Du, L.; Yang, T.; Wang, Y.; Guo, S.; Zhang, M.; Hussain, A.; et al. Core role of hydrophobic core of polymeric nanomicelle in endosomal escape of siRNA. Nano Lett. 2021, 21, 3680–3689. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yuk, S.A.; Dieterly, A.M.; Kwon, S.; Park, J.; Meng, F.; Gadalla, H.H.; Cadena, M.J.; Lyle, L.T.; Yeo, Y. Nanosac, a noncationic and soft polyphenol nanocapsule, enables systemic delivery of siRNA to solid tumors. ACS Nano 2021, 15, 4576–4593. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Liao, M.; Yu, S.; Yuan, B.; Jia, Z.; Zhou, L.; Tang, Y. Exosomes-delivered PD-L1 siRNA and CTLA-4 siRNA protect against growth and tumor immune escape in colorectal cancer. Genomics 2023, 115, 110646. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.F.A.; Kamaroddin, M.F.; Sajad, M. Cross-kingdom regulation by plant microRNAs provides novel insight into gene regulation. Adv. Nutr. 2021, 12, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, B. Plant-derived xenomiRs and cancer: Cross-kingdom gene regulation. Saudi J. Biol. Sci. 2021, 28, 2408–2422. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Z.; Zhou, Y.; Tong, Q.-S.; Duan, Q.-J.; Zhang, Q.; Du, J.-Z.; Yao, X.-Q. Precision medicine-guided co-delivery of ASPN siRNA and oxaliplatin by nanoparticles to overcome chemoresistance of colorectal cancer. Biomaterials 2022, 290, 121827. [Google Scholar] [CrossRef]
- Zou, Y.; Xiao, F.; Song, L.; Sun, B.; Sun, D.; Chu, D.; Wang, L.; Han, S.; Yu, Z.; O’Driscoll, C.M.; et al. A folate-targeted PEGylated cyclodextrin-based nanoformulation achieves co-delivery of docetaxel and siRNA for colorectal cancer. Int. J. Pharm. 2021, 606, 120888. [Google Scholar] [CrossRef]
- Okamoto, A.; Asai, T.; Hirai, Y.; Shimizu, K.; Koide, H.; Minamino, T.; Oku, N. Systemic administration of siRNA with anti-HB-EGF antibody-modified lipid nanoparticles for the treatment of triple-negative breast cancer. Mol. Pharm. 2018, 15, 1495–1504. [Google Scholar] [CrossRef]
- Chin, A.R.; Fong, M.Y.; Somlo, G.; Wu, J.; Swiderski, P.; Wu, X.; Wang, S.E. Cross-kingdom inhibition of breast cancer growth by plant miR159. Cell Res. 2016, 26, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Du, L.; Chen, Z.J.; Wang, X.; Ge, D.S.; Liu, N. LNP-miR-155 cy5 inhibitor regulates the copper transporter via the β-Catenin/TCF4/SLC31A1 signal for colorectal cancer therapy. Mol. Pharm. 2023, 20, 4138–4152. [Google Scholar] [CrossRef] [PubMed]
- Narasipura, E.A.; VanKeulen-Miller, R.; Ma, Y.T.; Fenton, O.S. Ongoing clinical trials of nonviral siRNA therapeutics. Bioconjugate Chem. 2023, 34, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, L.; Gu, Z.; Chen, Z.; Li, H.; Cheng, Z.; Li, H.; Zou, L. Co-delivery of microRNA-150 and quercetin by lipid nanoparticles (LNPs) for the targeted treatment of age-related macular degeneration (AMD). J. Control. Release 2023, 355, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Eygeris, Y.; Gupta, M.; Kim, J.; Sahay, G. Chemistry of lipid nanoparticles for RNA delivery. Acc. Chem. Res. 2022, 55, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Bui, T.A.; Yang, X.P.; Aksoy, Y.; Goldys, E.M.; Deng, W. Lipid-based nanoparticles for drug/gene delivery: An overview of the production techniques and difficulties encountered in their industrial development. ACS Mater. Au 2023, 3, 600–619. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xia, T. Recent advances in site-specific lipid nanoparticles for mRNA delivery. ACS Nanosci. Au 2023, 3, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhang, L.; Deng, W.; Lou, J.; Gao, X.; Lou, X.; Liu, Y.; Yao, X.; Sheng, Y.; Yan, Y.; et al. Spleen-selective co-delivery of mRNA and TLR4 agonist-loaded LNPs for synergistic immunostimulation and Th1 immune responses. J. Control. Release 2023, 357, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Aburai, K.; Hatanaka, K.; Takano, S.; Fujii, S.; Sakurai, K. Characterizing an siRNA-containing lipid-nanoparticle prepared by a microfluidic reactor: Small-angle X-ray scattering and cryotransmission electron microscopic studies. Langmuir 2020, 36, 12545–12554. [Google Scholar] [CrossRef]
- Henderson, M.I.; Eygeris, Y.; Jozic, A.; Herrera, M.; Sahay, G. Leveraging biological buffers for efficient messenger RNA delivery via lipid nanoparticles. Mol. Pharm. 2022, 19, 4275–4285. [Google Scholar] [CrossRef]
- El Moukhtari, S.H.; Garbayo, E.; Amundarain, A.; Pascual-Gil, S.; Carrasco-León, A.; Prosper, F.; Agirre, X.; Blanco-Prieto, M.J. Lipid nanoparticles for siRNA delivery in cancer treatment. J. Control. Release 2023, 361, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.M.S.; Chaudhary, N.; Arral, M.L.; Weiss, R.M.; Whitehead, K.A. The mixing method used to formulate lipid nanoparticles affects mRNA delivery efficacy and organ tropism. Eur. J. Pharm. Biopharm. 2023, 192, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Ickenstein, L.M.; Garidel, P. Lipid-based nanoparticle formulations for small molecules and RNA drugs. Expert. Opin. Drug Delivery 2019, 16, 1205–1226. [Google Scholar] [CrossRef] [PubMed]
- Khare, P.; Edgecomb, S.X.; Hamadani, C.M.; Tanner, E.E.; Manickam, D.S. Lipid nanoparticle-mediated drug delivery to the brain. Adv. Drug Deliv. Rev. 2023, 197, 114861. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.L.; Hajj, K.A.; Vizelman, J.; Bajaj, P.; Whitehead, K.A. Lipid nanoparticle formulations for enhanced co-delivery of siRNA and mRNA. Nano Lett. 2018, 18, 3814–3822. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Caracciolo, G. Looking back, moving forward: Lipid nanoparticles as a promising frontier in gene delivery. ACS Pharmacol. Transl. Sci. 2023, 6, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Jogdeo, C.M.; Panja, S.; Kanvinde, S.; Kapoor, E.; Siddhanta, K.; Oupický, D. Advances in Lipid-based codelivery systems for cancer and inflammatory diseases. Adv. Healthcare Mater. 2023, 12, 2202400. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, M.; Abazari, O.; Dayati, P.; Haghiralsadat, B.F.; Oroojalian, F.; Tofighi, D. Targeted delivery of 5-fluorouracil, miR-532-3p, and si-KRAS to the colorectal tumor using layer-by-layer liposomes. Front. Bioeng. Biotechnol. 2022, 10, 1013541. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, G.; Luo, X.; Wang, D.; Zhou, W.; Zhang, Y.; Zhang, W.; Chen, J.; Meng, Q.; Chen, E.; et al. Co-delivery of 5-fluorouracil and miRNA-34a mimics by host-guest self-assembly nanocarriers for efficacious targeted therapy in colorectal cancer patient-derived tumor xenografts. Theranostics 2021, 11, 2475–2489. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Day | Size (nm) | PDI | Zeta (mV) |
|---|---|---|---|---|
| LNP-VDX | 0 | 109.90 ± 0.26 | 0.101 ± 0.032 | 32.41 ± 1.23 |
| 15 | 124.00 ± 1.66 | 0.090 ± 0.006 | 35.79 ± 2.24 | |
| 30 | 125.07 ± 1.10 | 0.086 ± 0.012 | 36.09 ± 7.13 | |
| LNP-IRT | 0 | 133.80 ± 1.61 | 0.257 ± 0.010 | 31.14 ± 1.45 |
| 15 | 137.13 ± 2.84 | 0.162 ± 0.008 | 23.38 ± 0.59 | |
| 30 | 144.57 ± 1.08 | 0.233 ± 0.019 | 20.09 ± 0.33 | |
| LNP-I-V | 0 | 118.67 ± 1.27 | 0.171 ± 0.018 | 31.39 ± 1.64 |
| 15 | 135.47 ± 5.29 | 0.183 ± 0.060 | 21.98 ± 0.79 | |
| 30 | 154.10 ± 1.31 | 0.095 ± 0.043 | 34.70 ± 1.33 | |
| Blank LNP | 0 | 92.29 ± 0.57 | 0.214 ± 0.014 | 36.55 ± 1.83 |
| 15 | 108.93 ± 0.55 | 0.259 ± 0.002 | 21.59 ± 1.10 | |
| 30 | 268.23 ± 0.80 | 0.241 ± 0.025 | 41.57 ± 0.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, R.; Liu, Y.; Yang, N.; Zhang, T.; Hou, J.; He, Z.; Wang, Y.; Sun, X.; Shen, J.; Jiang, H.; et al. Combination of miR159 Mimics and Irinotecan Utilizing Lipid Nanoparticles for Enhanced Treatment of Colorectal Cancer. Pharmaceutics 2024, 16, 570. https://doi.org/10.3390/pharmaceutics16040570
Yang R, Liu Y, Yang N, Zhang T, Hou J, He Z, Wang Y, Sun X, Shen J, Jiang H, et al. Combination of miR159 Mimics and Irinotecan Utilizing Lipid Nanoparticles for Enhanced Treatment of Colorectal Cancer. Pharmaceutics. 2024; 16(4):570. https://doi.org/10.3390/pharmaceutics16040570
Chicago/Turabian StyleYang, Rulei, Yiran Liu, Ning Yang, Tian Zhang, Jiazhen Hou, Zongyan He, Yutong Wang, Xujie Sun, Jingshan Shen, Hualiang Jiang, and et al. 2024. "Combination of miR159 Mimics and Irinotecan Utilizing Lipid Nanoparticles for Enhanced Treatment of Colorectal Cancer" Pharmaceutics 16, no. 4: 570. https://doi.org/10.3390/pharmaceutics16040570