Evaluation of the Transport and Binding of Dopamine-Loaded PLGA Nanoparticles for the Treatment of Parkinson’s Disease Using In Vitro Model Systems

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Nanoparticle Synthesis and Characterization
2.1.1. Nanoparticle Synthesis
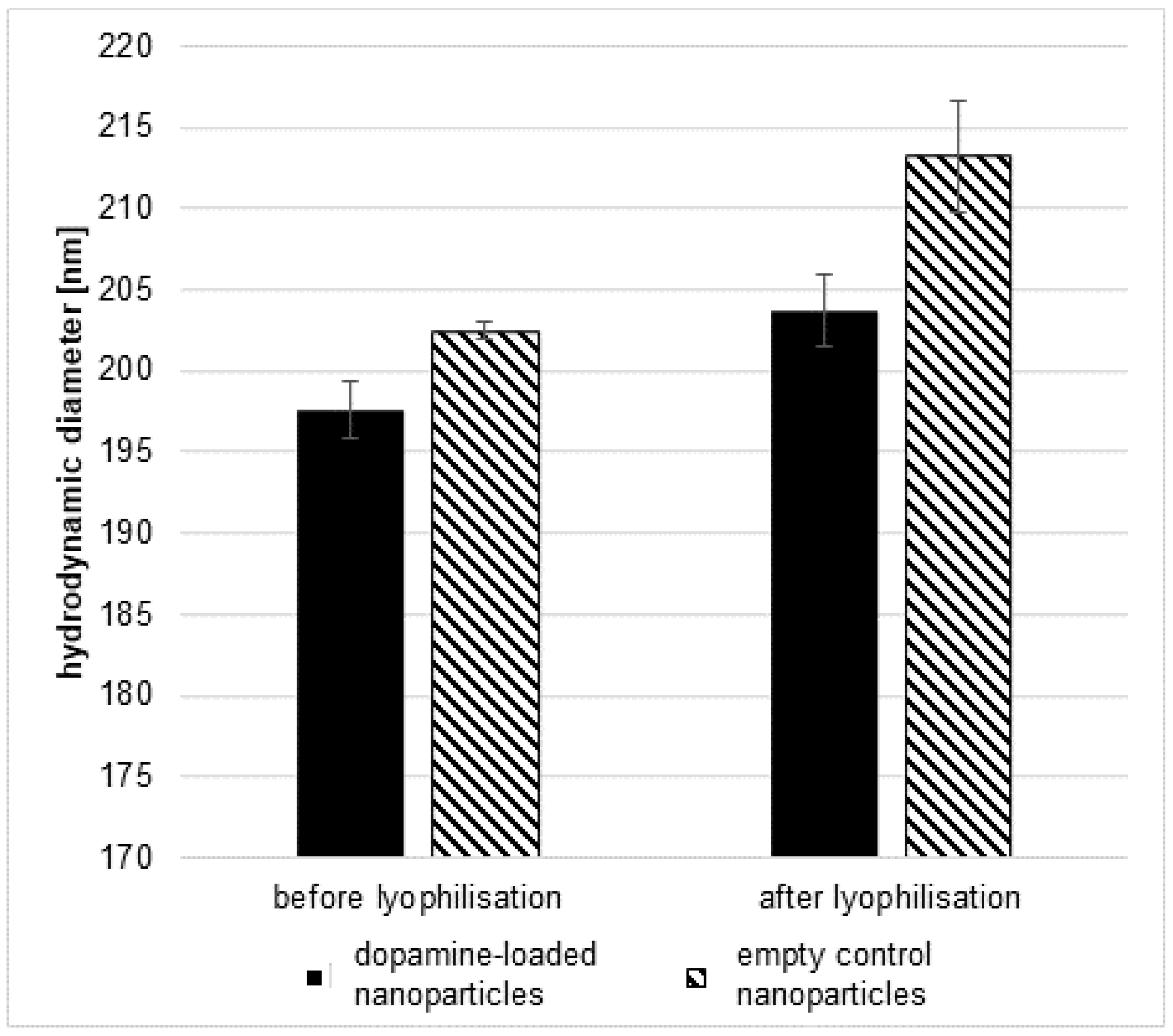
2.1.2. Lyophilisation of Nanoparticles
2.1.3. Water Content Determination
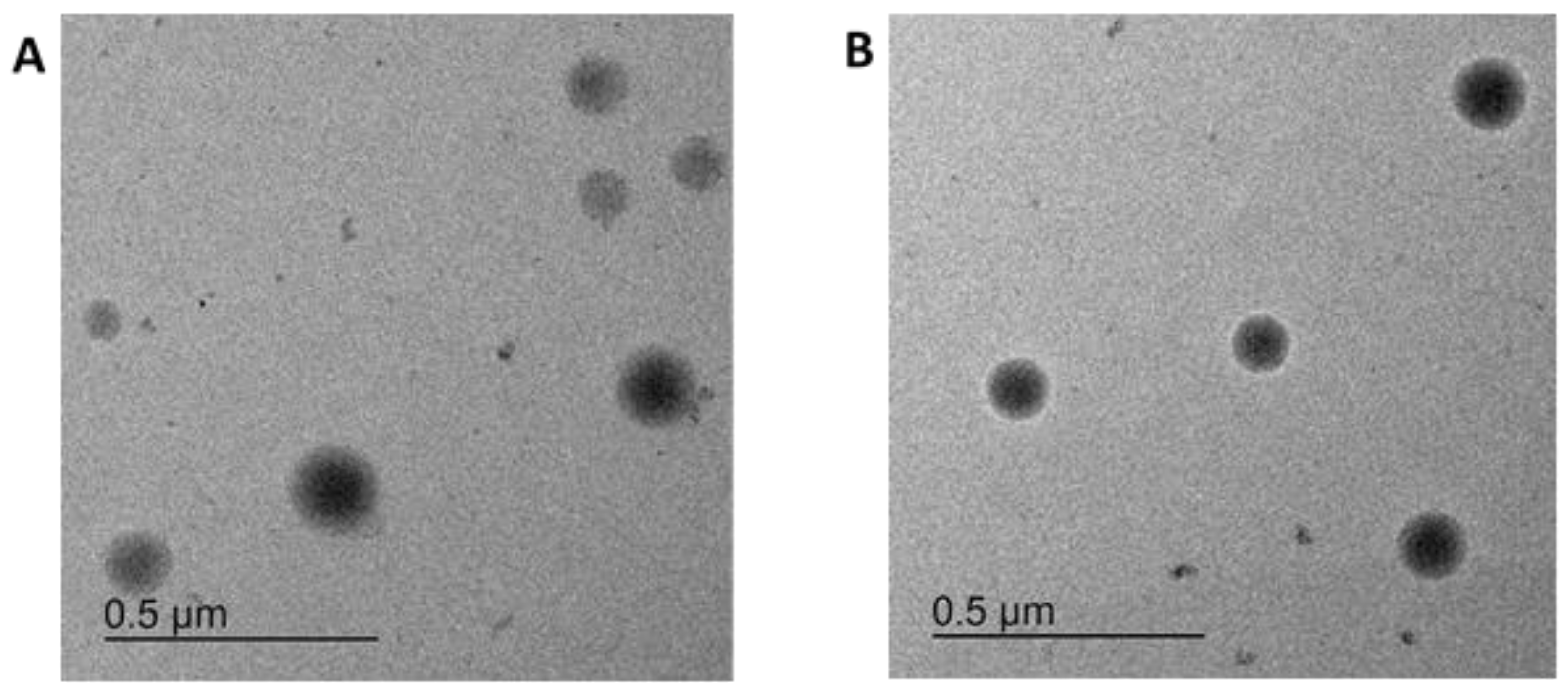
2.1.4. Particle Characterisation
2.1.5. Nanoparticle Leaching
2.2. Cell Culture
2.2.1. Cell Lines
2.2.2. hiPSC-Derived Human Brain Capillary Endothelial Cells
2.2.3. Cytotoxicity Testing
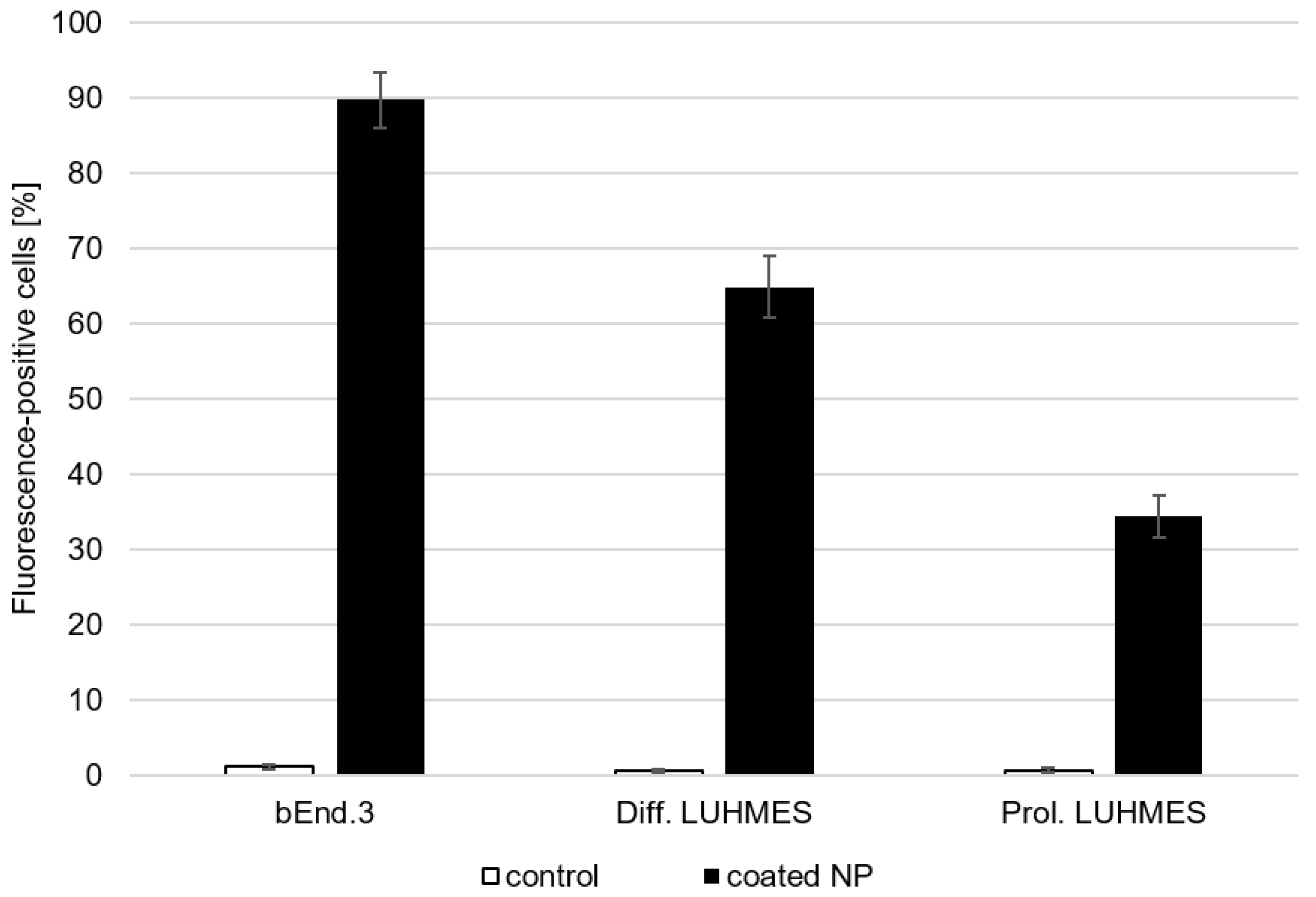
2.2.4. Binding and Uptake Studies
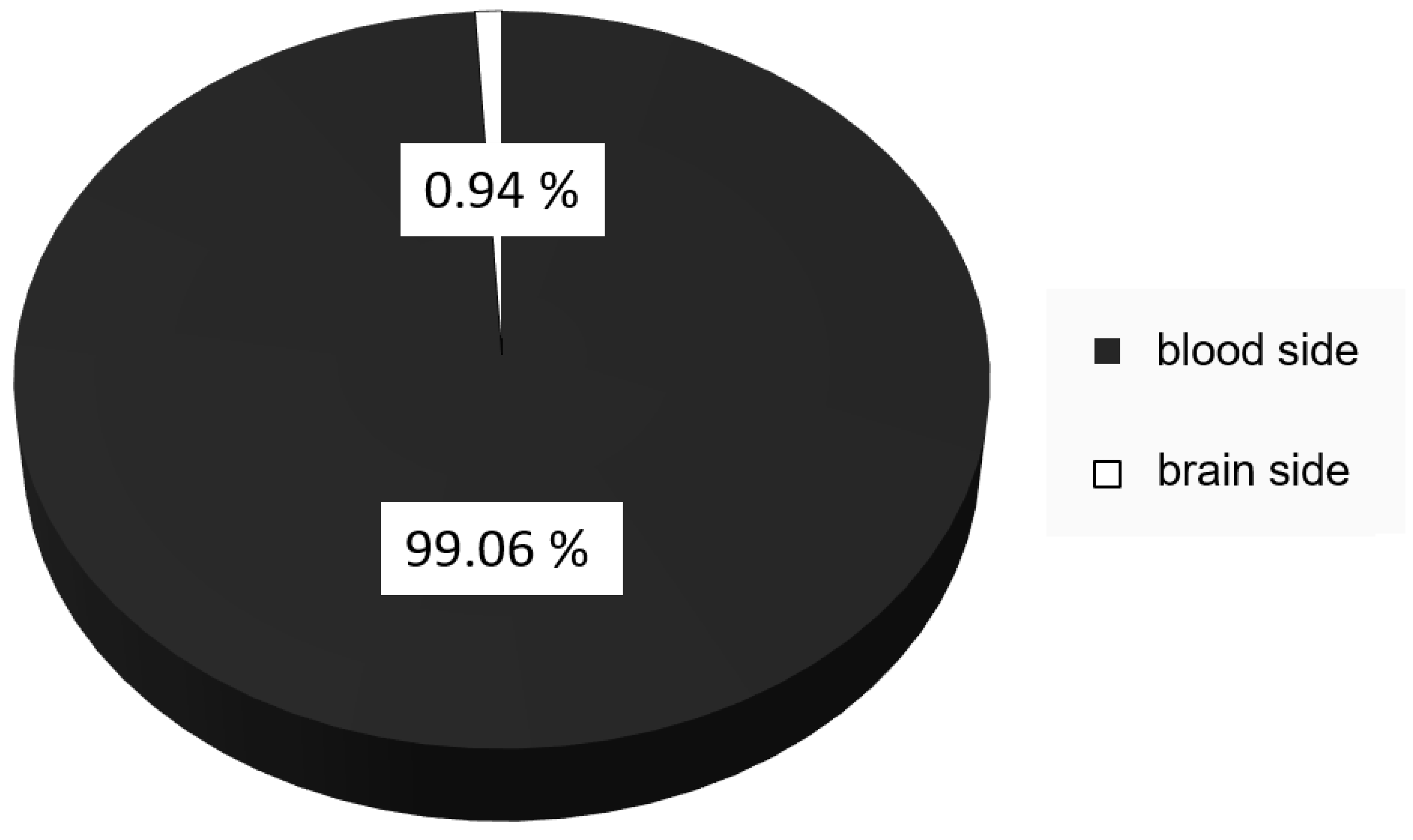
2.2.5. Transport Studies
2.2.6. Dopamine Quantification
2.3. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darweesh, S.K.; Koudstaal, P.J.; Stricker, B.H.; Hofman, A.; Ikram, M.A. Trends in the Incidence of Parkinson Disease in the General Population: The Rotterdam Study. Am. J. Epidemiol. 2016, 183, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Rocca, W.A. Time Trends in the Incidence of Parkinson Disease. JAMA Neurol. 2016, 73, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A. Presynaptic Mechanisms of l-DOPA-Induced Dyskinesia: The Findings, the Debate, and the Therapeutic Implications. Front. Neurol. 2014, 5, 242. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Lianeri, M.; Kozubski, W. Molecular Effects of L-dopa Therapy in Parkinson’s Disease. Curr. Genom. 2014, 15, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Poupot, R.; Bergozza, D.; Fruchon, S. Nanoparticle-Based Strategies to Treat Neuro-Inflammation. Materials 2018, 11, 270. [Google Scholar] [CrossRef]
- Hasannejad-Asl, B.; Pooresmaeil, F.; Choupani, E.; Dabiri, M.; Behmardi, A.; Fadaie, M.; Fathi, M.; Moosavi, S.A.; Takamoli, S.; Hemati, E.; et al. Nanoparticles as Powerful Tools for Crossing the Blood-brain Barrier. CNS Neurol. Disord. Drug Targets 2023, 22, 18–26. [Google Scholar]
- Bazi Alahri, M.; Jibril Ibrahim, A.; Barani, M.; Arkaban, H.; Shadman, S.M.; Salarpour, S.; Zarrintaj, P.; Jaberi, J.; Turki Jalil, A. Management of Brain Cancer and Neurodegenerative Disorders with Polymer-Based Nanoparticles as a Biocompatible Platform. Molecules 2023, 28, 841. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Silva, V.; Barreiro, S.; Silva, R.; Remião, F.; Borges, F.; Fernandes, C. Brain drug delivery and neurodegenerative diseases: Polymeric PLGA-based nanoparticles as a forefront platform. Ageing Res. Rev. 2022, 79, 101658. [Google Scholar] [CrossRef] [PubMed]
- Pahuja, R.; Seth, K.; Shukla, A.; Shukla, R.K.; Bhatnagar, P.; Chauhan, L.K.; Saxena, P.N.; Arun, J.; Chaudhari, B.P.; Patel, D.K.; et al. Trans-blood brain barrier delivery of dopamine-loaded nanoparticles reverses functional deficits in parkinsonian rats. ACS Nano 2015, 9, 4850–4871. [Google Scholar] [CrossRef]
- Shin, M.; Kim, H.K.; Lee, H. Dopamine-loaded poly(D,L-lactic-co-glycolic acid) microspheres: New strategy for encapsulating small hydrophilic drugs with high efficiency. Biotechnol. Prog. 2014, 30, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A.; De Giglio, E.; Cafagna, D.; Denora, N.; Agrimi, G.; Cassano, T.; Gaetani, S.; Cuomo, V.; Trapani, G. Characterization and evaluation of chitosan nanoparticles for dopamine brain delivery. Int. J. Pharm. 2011, 419, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Bicker, J.; Alves, G.; Fortuna, A.; Falcão, A. Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: A review. Eur. J. Pharm. Biopharm. 2014, 87, 409–432. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Shi, Y.; Azevedo, H.S. In vitro blood-brain barrier models for drug research: State-of-the-art and new perspectives on reconstituting these models on artificial basement membrane platforms. Drug Discov. Today 2016, 21, 1367–1386. [Google Scholar] [CrossRef]
- Jain, P.; Pawar, R.S.; Pandey, R.S.; Madan, J.; Pawar, S.; Lakshmi, P.K.; Sudheesh, M.S. In-vitro in-vivo correlation (IVIVC) in nanomedicine: Is protein corona the missing link? Biotechnol. Adv. 2017, 35, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, E.S.; Al-Ahmad, A.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 2014, 4, 4160. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. Commentary on human pluripotent stem cell-based blood-brain barrier models. Fluids Barriers CNS 2020, 17, 64. [Google Scholar] [CrossRef]
- Roux, G.L.; Jarray, R.; Guyot, A.C.; Pavoni, S.; Costa, N.; Théodoro, F.; Nassor, F.; Pruvost, A.; Tournier, N.; Kiyan, Y.; et al. Proof-of-Concept Study of Drug Brain Permeability Between in Vivo Human Brain and an in Vitro iPSCs-Human Blood-Brain Barrier Model. Sci. Rep. 2019, 9, 16310. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Zensi, A.; Wien, S.L.; Tschickardt, S.E.; Maier, W.; Vogel, T.; Worek, F.; Pietrzik, C.U.; Kreuter, J.; von Briesen, H. Uptake mechanism of ApoE-modified nanoparticles on brain capillary endothelial cells as a blood-brain barrier model. PLoS ONE 2012, 7, e32568. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J. Drug Target 2002, 10, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Zensi, A.; Begley, D.; Pontikis, C.; Legros, C.; Mihoreanu, L.; Wagner, S.; Büchel, C.; von Briesen, H.; Kreuter, J. Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J. Control Release 2009, 137, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Kleimann, P. Der Einfluss von Nanopartikelstabilisatoren auf die Flüssigkeitszerstäubung mittels Druckluft-, Ultraschall- und Lochmembranverneblern. Ph.D. Dissertation, Justus-Liebig-Universität Gießen, Gießen, Germany, 4 November 2014. [Google Scholar]
- Margreth, M.; Schlink, R.; Steinbach, A. Water Determination By Karl Fischer Titration. In Pharmaceutical Sciences Encyclopedia: Drug Discovery, Development, and Manufacturing; Gad, S.C., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef]
- Steeg, R.; Neubauer, J.C.; Müller, S.C.; Ebneth, A.; Zimmermann, H. The EBiSC iPSC bank for disease studies. Stem Cell Res. 2020, 49, 102034. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Pöltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef]
- Stebbins, M.J.; Wilson, H.K.; Canfield, S.G.; Qian, T.; Palecek, S.P.; Shusta, E.V. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods 2016, 101, 93–102. [Google Scholar] [CrossRef]
- Danz, K.; Höcherl, T.; Wien, S.L.; Wien, L.; von Briesen, H.; Wagner, S. Experimental Comparison of Primary and hiPS-Based In Vitro Blood-Brain Barrier Models for Pharmacological Research. Pharmaceutics 2022, 14, 737. [Google Scholar] [CrossRef]
- Gelperina, S.; Maksimenko, O.; Khalansky, A.; Vanchugova, L.; Shipulo, E.; Abbasova, K.; Berdiev, R.; Wohlfart, S.; Chepurnova, N.; Kreuter, J. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: Influence of the formulation parameters. Eur. J. Pharm. Biopharm. 2010, 74, 157–163. [Google Scholar] [CrossRef]
- Wagner, S.; Kufleitner, J.; Zensi, A.; Dadparvar, M.; Wien, S.; Bungert, J.; Vogel, T.; Worek, F.; Kreuter, J.; von Briesen, H. Nanoparticulate transport of oximes over an in vitro blood-brain barrier model. PLoS ONE 2010, 5, e14213. [Google Scholar] [CrossRef]
- Pardridge, W.M. Advances in cell biology of blood-brain barrier transport. Semin. Cell Biol. 1991, 2, 419–426. [Google Scholar]
- Kreuter, J.; Gelperina, S. Use of nanoparticles for cerebral cancer. Tumori 2008, 94, 271–277. [Google Scholar] [CrossRef]
- Cohen-Sela, E.; Chorny, M.; Koroukhov, N.; Danenberg, H.D.; Golomb, G. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J. Control Release 2009, 133, 90–95. [Google Scholar] [CrossRef]
- Barichello, J.M.; Morishita, M.; Takayama, K.; Nagai, T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev. Ind. Pharm. 1999, 25, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Govender, T.; Stolnik, S.; Garnett, M.C.; Illum, L.; Davis, S.S. PLGA nanoparticles prepared by nanoprecipitation: Drug loading and release studies of a water soluble drug. J. Control Release 1999, 57, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Leyva-Gómez, G.; Cortés, H.; Magaña, J.J.; Leyva-García, N.; Quintanar-Guerrero, D.; Florán, B. Nanoparticle technology for treatment of Parkinson’s disease: The role of surface phenomena in reaching the brain. Drug Discov. Today 2015, 20, 824–837. [Google Scholar] [CrossRef]
- Huile, G.; Shuaiqi, P.; Zhi, Y.; Shijie, C.; Chen, C.; Xinguo, J.; Shun, S.; Zhiqing, P.; Yu, H. A cascade targeting strategy for brain neuroglial cells employing nanoparticles modified with angiopep-2 peptide and EGFP-EGF1 protein. Biomaterials 2011, 32, 8669–8675. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Ma, H.; Guo, Y.; Liu, S.; Kuang, Y.; Shao, K.; Li, J.; Liu, Y.; Han, L.; Huang, S.; et al. Angiopep-conjugated nanoparticles for targeted long-term gene therapy of Parkinson’s disease. Pharm. Res. 2013, 30, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Stab, J.; Zlatev, I.; Raudszus, B.; Meister, S.; Pietrzik, C.U.; Langer, K.; von Briesen, H.; Wagner, S. Flurbiprofen-loaded Nanoparticles can Cross a Primary Porcine In vitro Blood-Brain Barrier Model to Reduce Amyloid- B 42 Burden. J. Nanomed. Biother. Discov. 2016, 6, 1–10. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrodynamic Diameter [nm] | PDI | Zeta Potential [mV] | Dopamine Content [µg/mg NP] | |
|---|---|---|---|---|
| with dopamine | 198.1 ± 3.4 | 0.03 ± 0.01 | −29.1 ± 1.7 | 4.1 ± 0.4 |
| without dopamine | 207.5 ± 5.5 | 0.04 ± 0.02 | −33.8 ± 2.5 | — |
| Particle Type | TEM | NTA | Zeta Potential |
|---|---|---|---|
| particle with dopamine and with Lumogen Red 305 dye | 203 ± 44 nm | 187 ± 55 nm | −5 ± 1 mV |
| particle with Lumogen Red 305 dye | 167 ± 63 nm | 184 ± 52 nm | −5 ± 1 mV |
| Particle Type | Fluorescence of the Filtrate [%] at t = 0 h | Fluorescence of the Filtrate [%] at t = 24 h |
|---|---|---|
| particle with dopamine and with Lumogen Red 305 dye | 4.8 ± 0.1 | 5.6 ± 3.6 |
| particle with Lumogen Red 305 dye | 6.2 ± 0.3 | 7.4 ± 2.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danz, K.; Fleddermann, J.; Koch, M.; Fecioru, E.; Maahs, L.; Kinsinger, N.; Krämer, J.; Kraegeloh, A.; Wagner, S. Evaluation of the Transport and Binding of Dopamine-Loaded PLGA Nanoparticles for the Treatment of Parkinson’s Disease Using In Vitro Model Systems. Pharmaceutics 2024, 16, 571. https://doi.org/10.3390/pharmaceutics16050571
Danz K, Fleddermann J, Koch M, Fecioru E, Maahs L, Kinsinger N, Krämer J, Kraegeloh A, Wagner S. Evaluation of the Transport and Binding of Dopamine-Loaded PLGA Nanoparticles for the Treatment of Parkinson’s Disease Using In Vitro Model Systems. Pharmaceutics. 2024; 16(5):571. https://doi.org/10.3390/pharmaceutics16050571
Chicago/Turabian StyleDanz, Karin, Jana Fleddermann, Marcus Koch, Elena Fecioru, Lorenz Maahs, Nicole Kinsinger, Johannes Krämer, Annette Kraegeloh, and Sylvia Wagner. 2024. "Evaluation of the Transport and Binding of Dopamine-Loaded PLGA Nanoparticles for the Treatment of Parkinson’s Disease Using In Vitro Model Systems" Pharmaceutics 16, no. 5: 571. https://doi.org/10.3390/pharmaceutics16050571