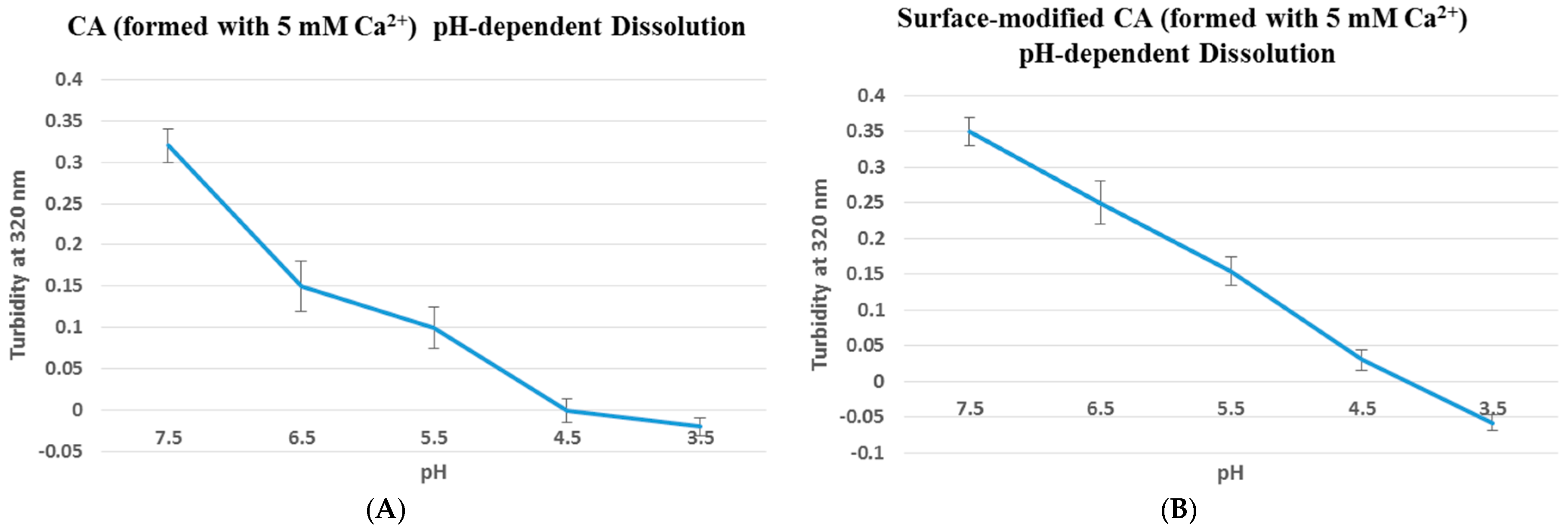
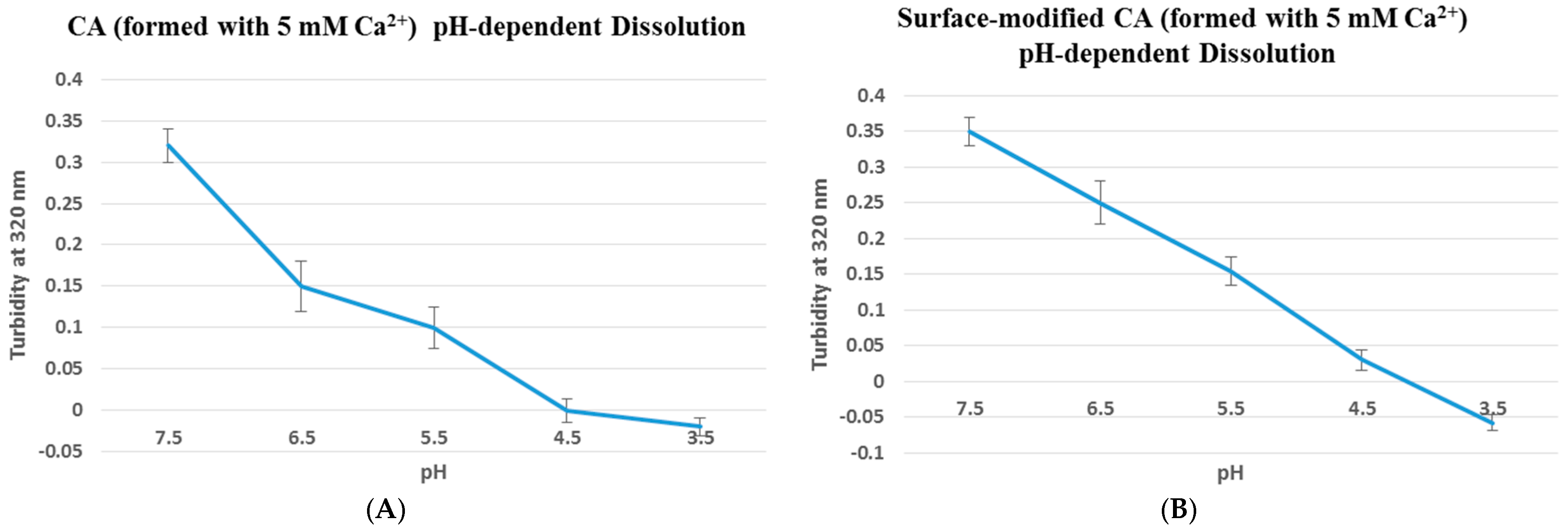
Figure 1.
pH dissolution test was done using UV spectrophotometer at 320 nm wavelength. Particles without (A) or with (B) surface-coating (streptavidin–biotin PEG–fibronectin) were initially fabricated in 100 µL of bicarbonate-buffered DMEM media (pH 7.5), followed by addition of DMEM media of decreasing pHs (6.5, 5.5, 4.5 and 3.5) to top up to 1 mL, prior to the turbidity measurement. Turbidity value of 0.36 indicates that particles were already formed at pH 7.5, and with decreasing pHs the particles started to dissolve, resulting in lower turbidity value until reaching close to 0 where all particles were completely dissolved.
Figure 1.
pH dissolution test was done using UV spectrophotometer at 320 nm wavelength. Particles without (A) or with (B) surface-coating (streptavidin–biotin PEG–fibronectin) were initially fabricated in 100 µL of bicarbonate-buffered DMEM media (pH 7.5), followed by addition of DMEM media of decreasing pHs (6.5, 5.5, 4.5 and 3.5) to top up to 1 mL, prior to the turbidity measurement. Turbidity value of 0.36 indicates that particles were already formed at pH 7.5, and with decreasing pHs the particles started to dissolve, resulting in lower turbidity value until reaching close to 0 where all particles were completely dissolved.
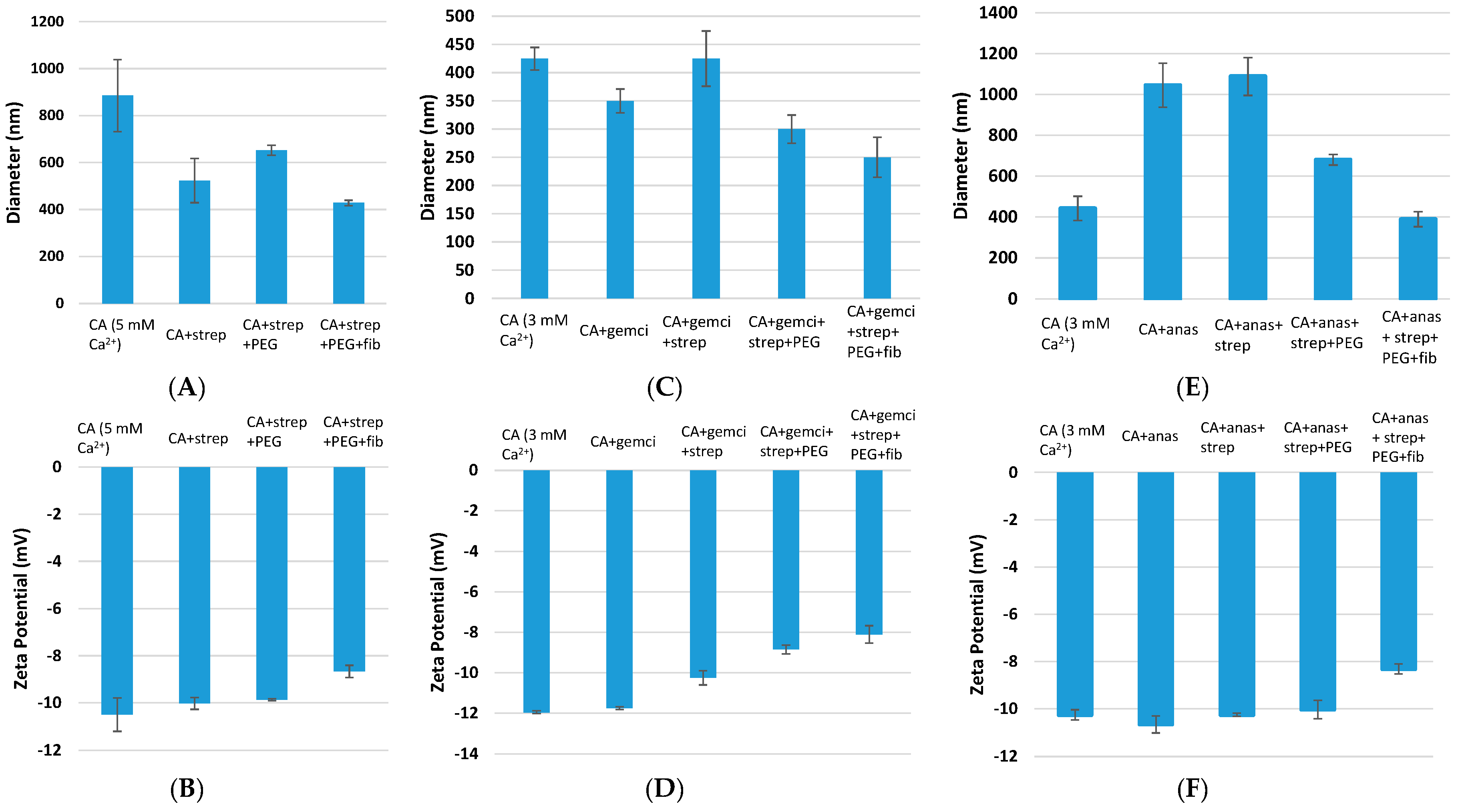
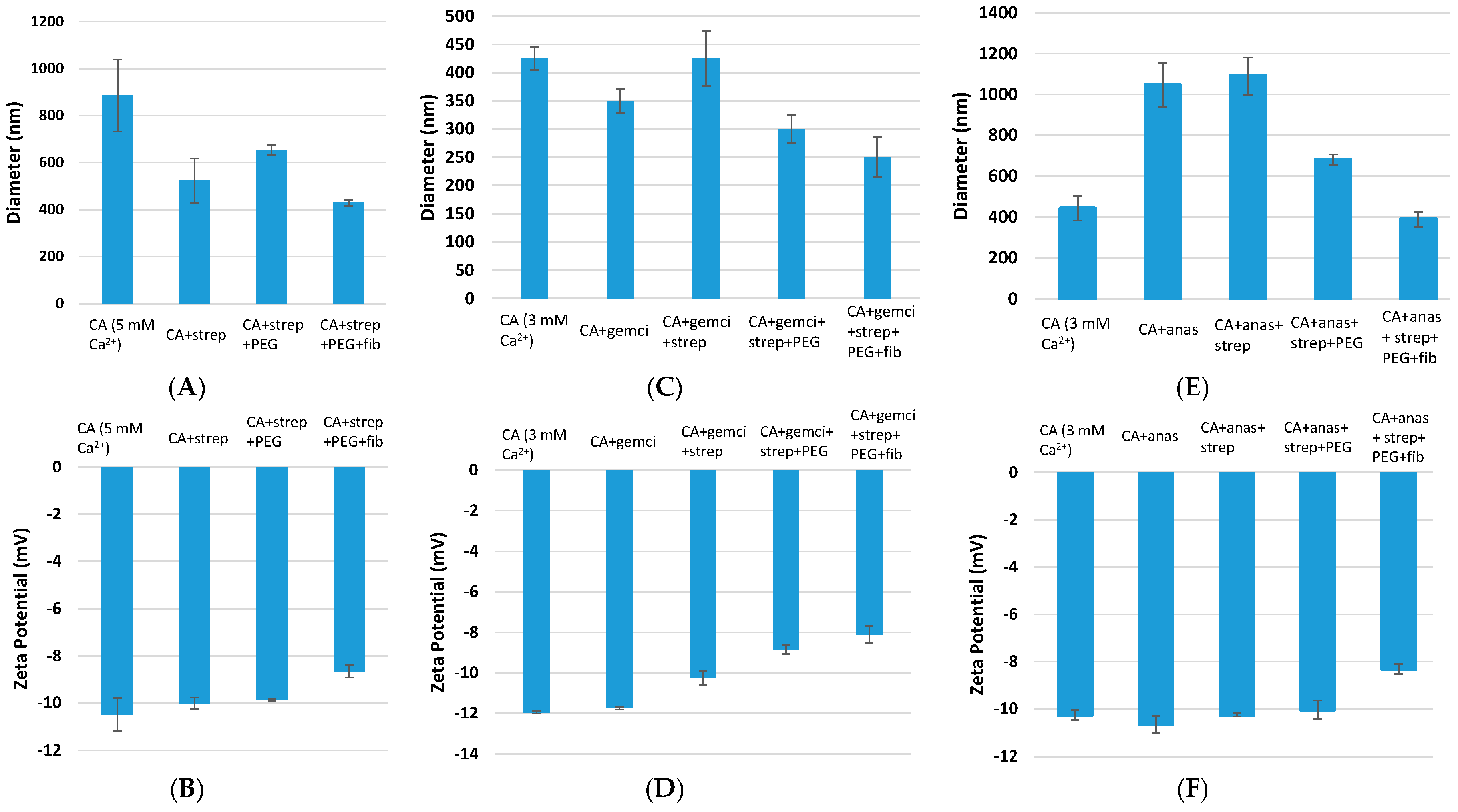
Figure 2.
Size and zeta potential measurement of surface-modified, drug-loaded CA particles. CA (fabricated with 3 mM Ca2+) and CA + drug (1 μM drug concentration) were used as controls. Surface modifications included: CA + drug + streptavidin, CA + drug + streptavidin + biotin–PEG, and CA + drug + streptavidin + biotin–PEG + fibronectin. CA: carbonate apatite; strep: streptavidin; fib: fibronectin; gemci: gemcitabine; anas: anastrozole. (A) and (B) denote particle diameter and surface charge measurements for unmodified and surface-modified particles without incorporated drugs, (C) and (D) denote particle diameter and surface charge measurements for unmodified and surface-modified particles with incorporated gemcitabine, and (E) and (F) denote particle diameter and surface charge measurements for unmodified and surface-modified particles with incorporated anastrozole.
Figure 2.
Size and zeta potential measurement of surface-modified, drug-loaded CA particles. CA (fabricated with 3 mM Ca2+) and CA + drug (1 μM drug concentration) were used as controls. Surface modifications included: CA + drug + streptavidin, CA + drug + streptavidin + biotin–PEG, and CA + drug + streptavidin + biotin–PEG + fibronectin. CA: carbonate apatite; strep: streptavidin; fib: fibronectin; gemci: gemcitabine; anas: anastrozole. (A) and (B) denote particle diameter and surface charge measurements for unmodified and surface-modified particles without incorporated drugs, (C) and (D) denote particle diameter and surface charge measurements for unmodified and surface-modified particles with incorporated gemcitabine, and (E) and (F) denote particle diameter and surface charge measurements for unmodified and surface-modified particles with incorporated anastrozole.
Figure 3.
After formation of surface-modified CA as described above, centrifugation was done at 4 °C with 13,000 rpm for 15 min. The supernatant was discarded and the pellet was resuspended at 100 μL DMEM. Six microliters of samples and loading dye with 1:1 ratio were loaded into each gel well (BioRad Precast Gels 7.5%) and run through SDS PAGE at 60 V for 1 h. The resulting gel was processed through silver staining.
Figure 3.
After formation of surface-modified CA as described above, centrifugation was done at 4 °C with 13,000 rpm for 15 min. The supernatant was discarded and the pellet was resuspended at 100 μL DMEM. Six microliters of samples and loading dye with 1:1 ratio were loaded into each gel well (BioRad Precast Gels 7.5%) and run through SDS PAGE at 60 V for 1 h. The resulting gel was processed through silver staining.
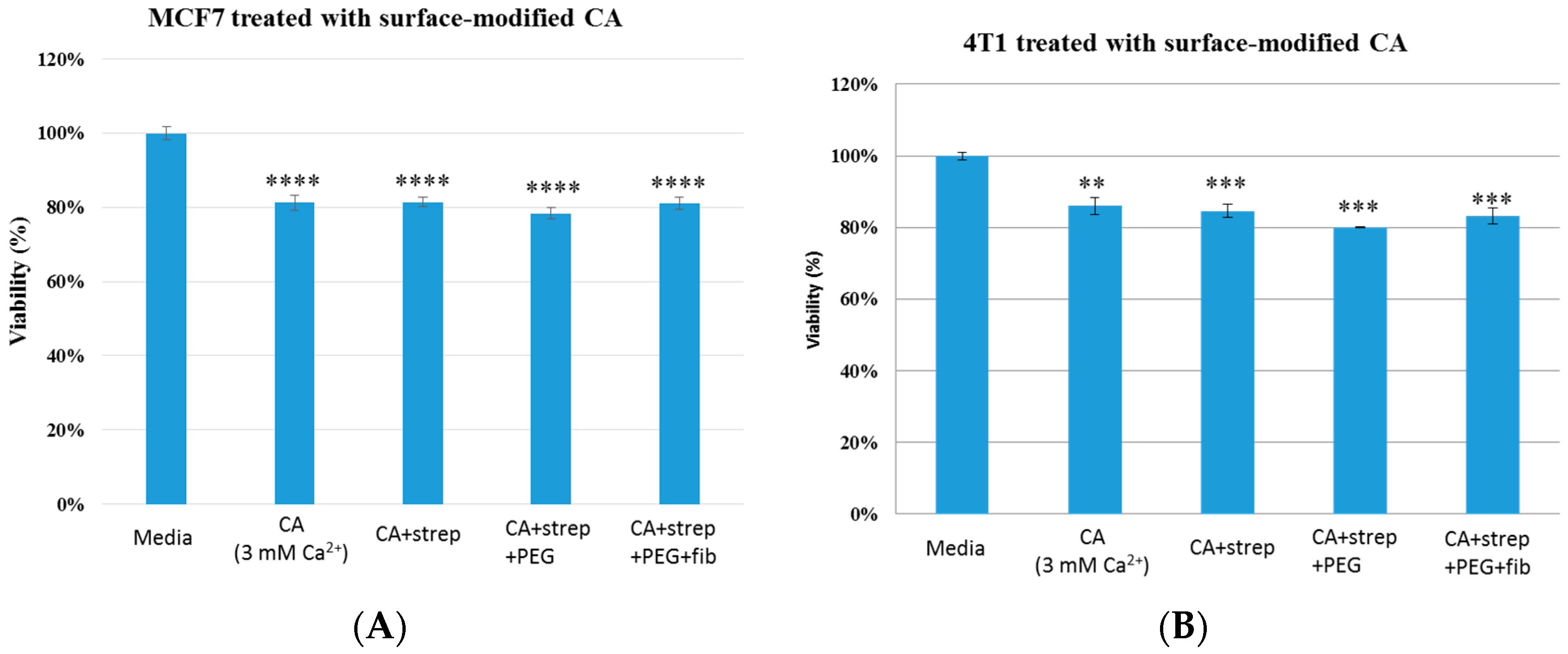
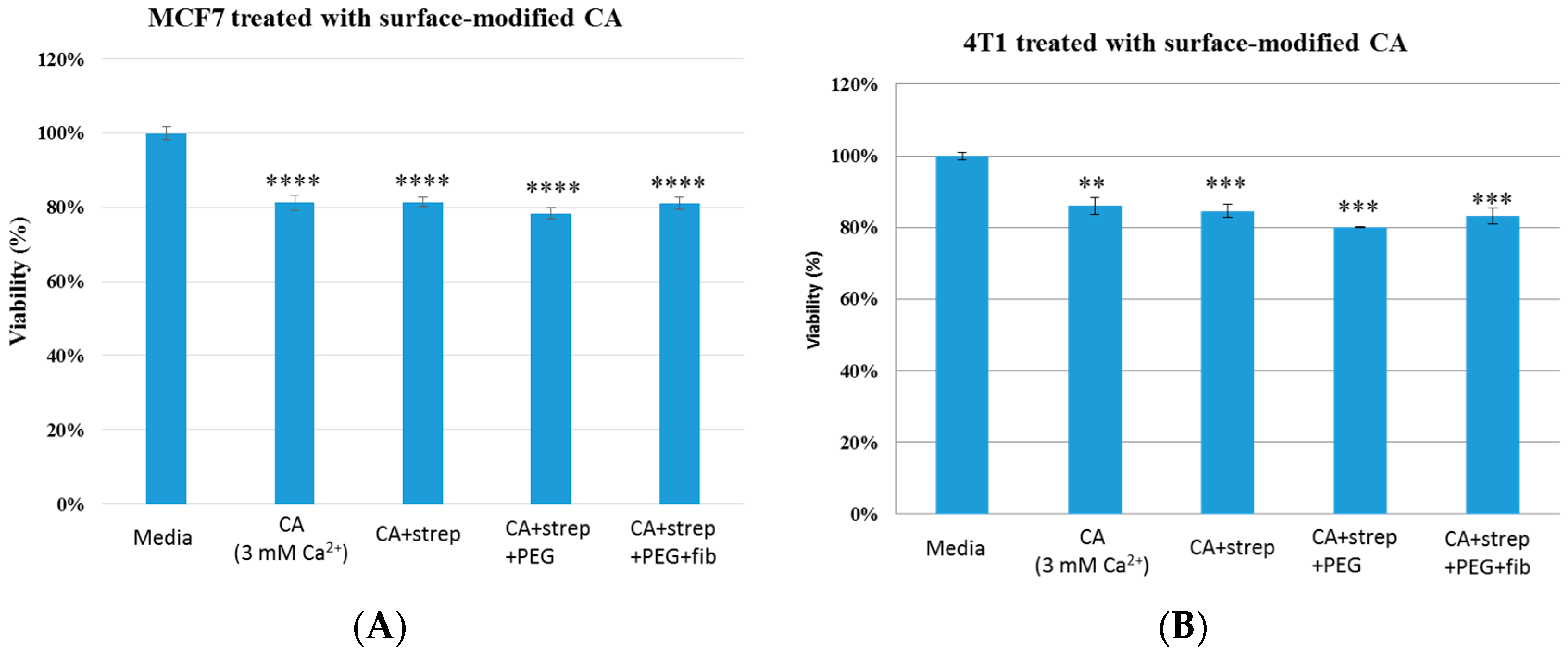
Figure 4.
Cell viability of MCF7 (A) and 4T1 (B) cells after 48 h of treatment with CA and surface- modified CA particles. Surface modifications includes CA + streptavidin, CA + streptavidin + biotin–PEG and CA + streptavidin + biotin–PEG + fibronectin with untreated cells and CA particles alone considered as controls. CA: carbonate apatite; strep: streptavidin; fib: fibronectin.
Figure 4.
Cell viability of MCF7 (A) and 4T1 (B) cells after 48 h of treatment with CA and surface- modified CA particles. Surface modifications includes CA + streptavidin, CA + streptavidin + biotin–PEG and CA + streptavidin + biotin–PEG + fibronectin with untreated cells and CA particles alone considered as controls. CA: carbonate apatite; strep: streptavidin; fib: fibronectin.
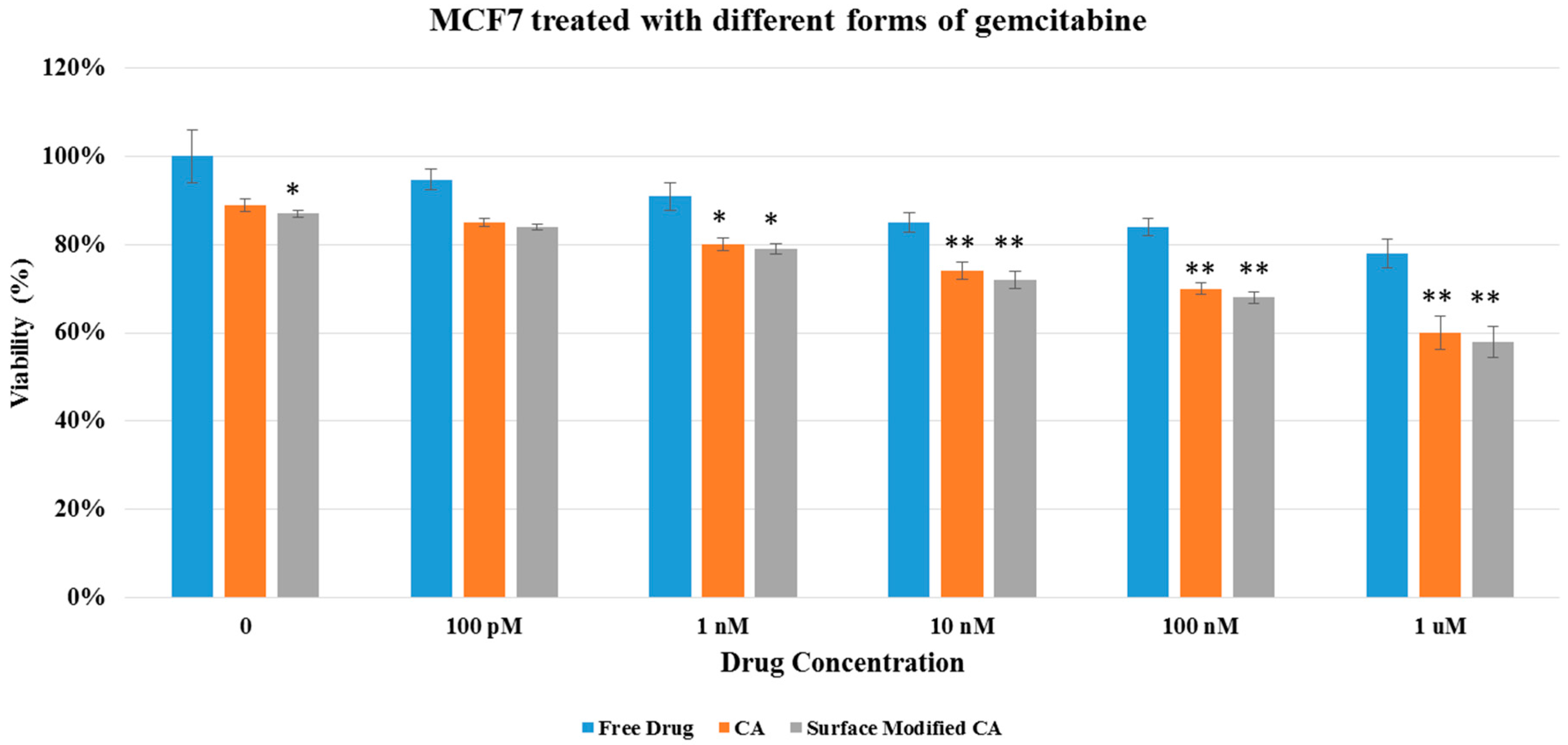
Figure 5.
Cell viability of MCF7 after 48 h of treatment with gemcitabine-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were significant (*) at p value 0.01 to 0.05 and very significant (**) at p value 0.001 to 0.01.
Figure 5.
Cell viability of MCF7 after 48 h of treatment with gemcitabine-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were significant (*) at p value 0.01 to 0.05 and very significant (**) at p value 0.001 to 0.01.
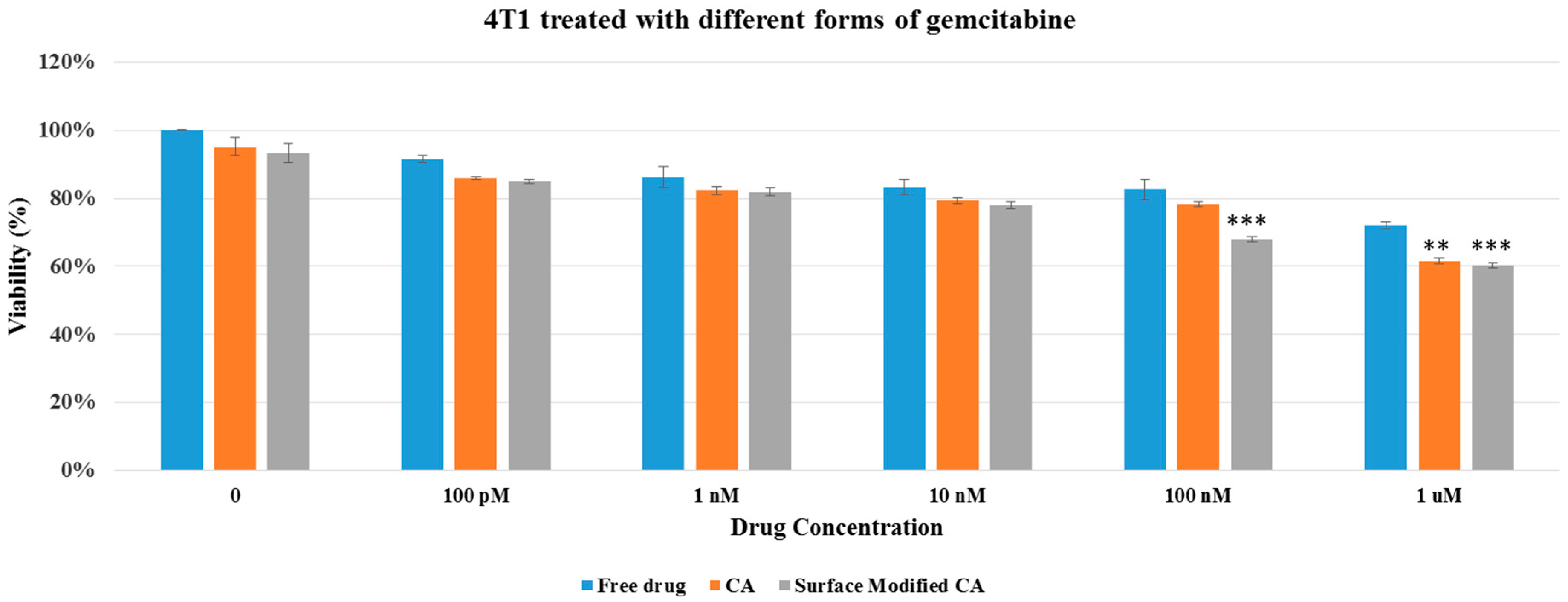
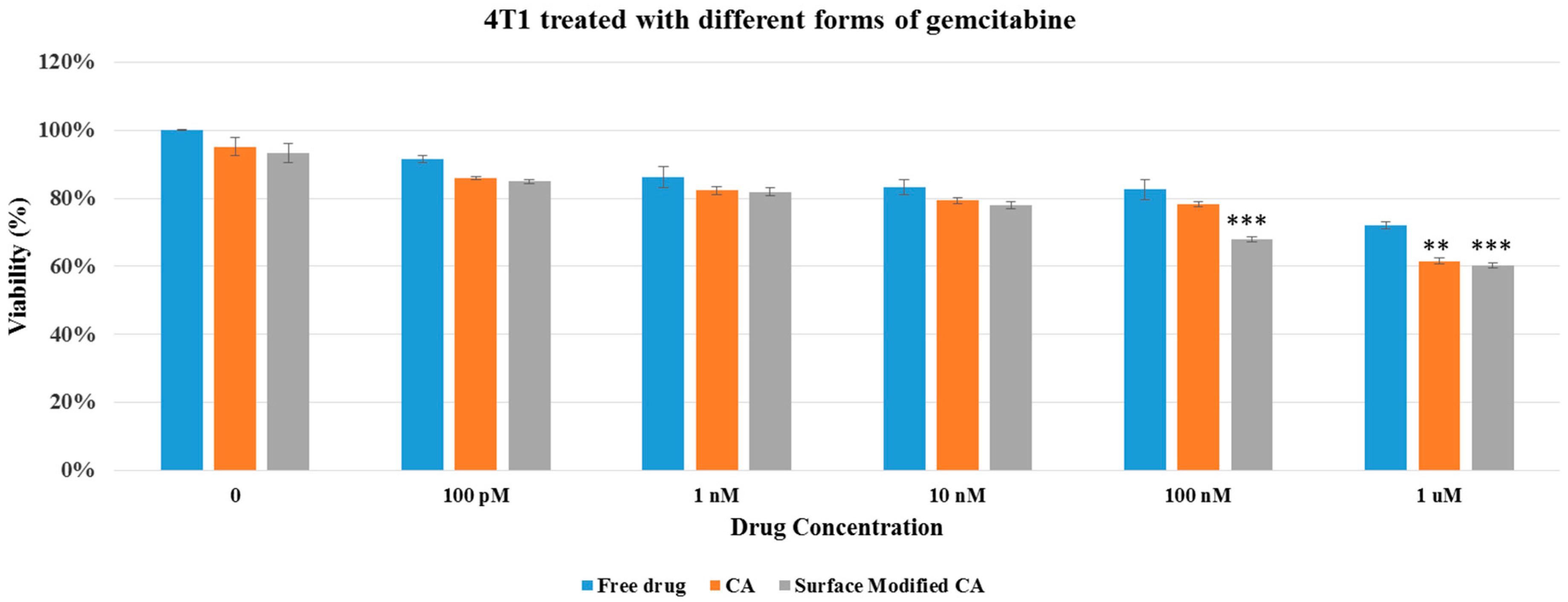
Figure 6.
Cell viability of 4T1 after 48 h of treatment with gemcitabine-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were very significant (**) at p value 0.001 to 0.01, and extremely significant (***) at p value 0.0001 to 0.001.
Figure 6.
Cell viability of 4T1 after 48 h of treatment with gemcitabine-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were very significant (**) at p value 0.001 to 0.01, and extremely significant (***) at p value 0.0001 to 0.001.
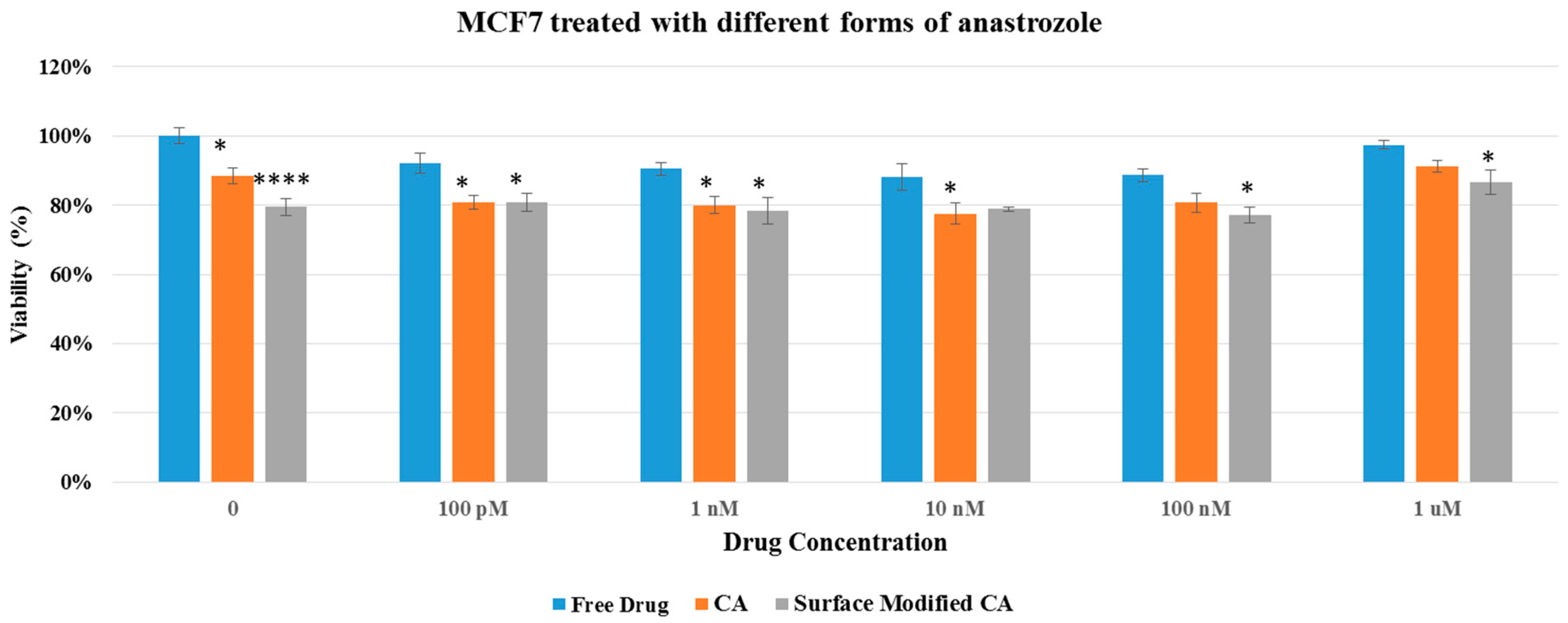
Figure 7.
Cell viability of MCF7 after 48 h of treatment with anastrozole-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were significant (*) at p value 0.01 to 0.05, very significant (**) at p value 0.001 to 0.01, extremely significant (***) at p value 0.0001 to 0.001, and extremely significant (****) at p value < 0.0001.
Figure 7.
Cell viability of MCF7 after 48 h of treatment with anastrozole-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were significant (*) at p value 0.01 to 0.05, very significant (**) at p value 0.001 to 0.01, extremely significant (***) at p value 0.0001 to 0.001, and extremely significant (****) at p value < 0.0001.
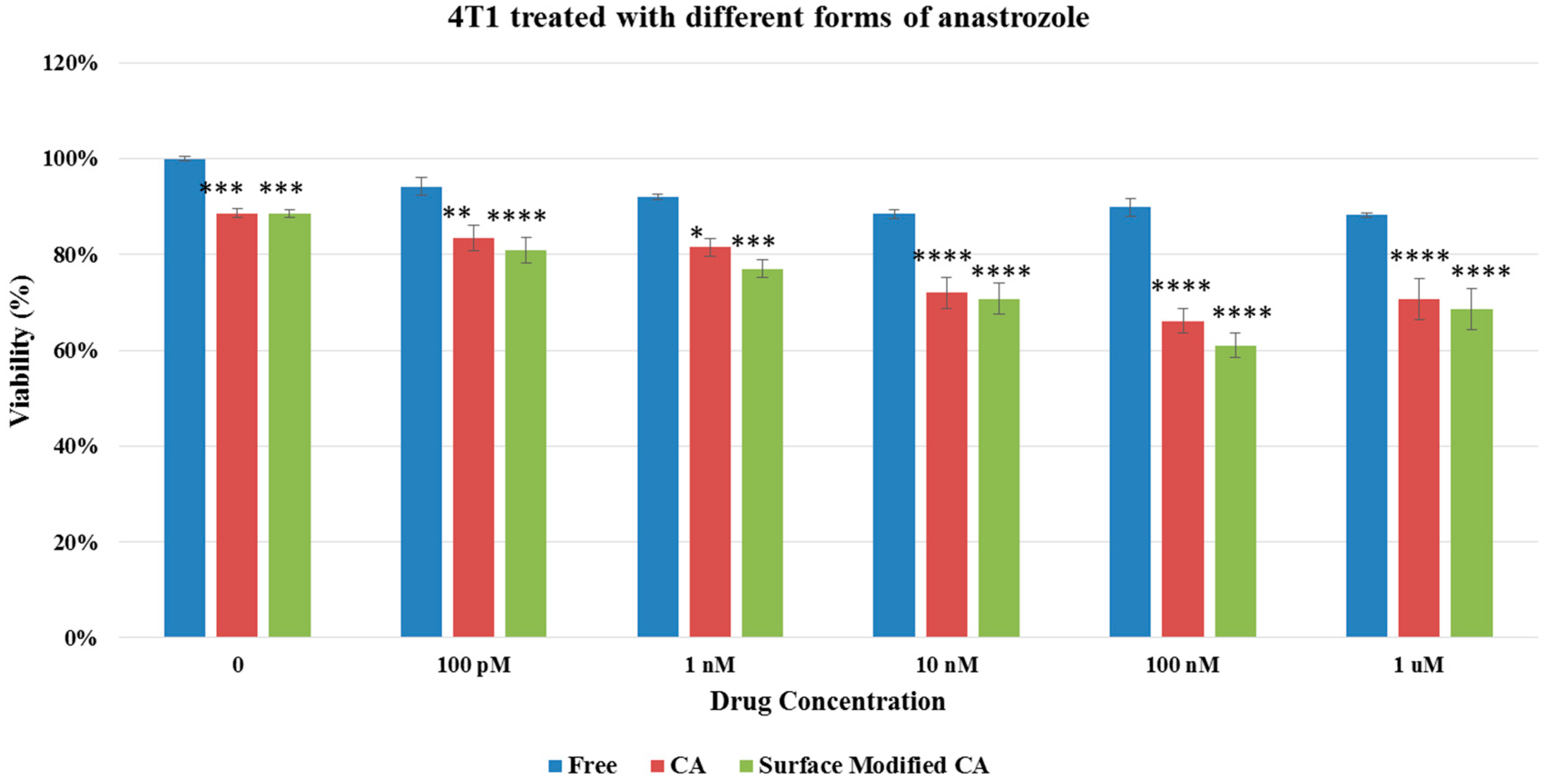
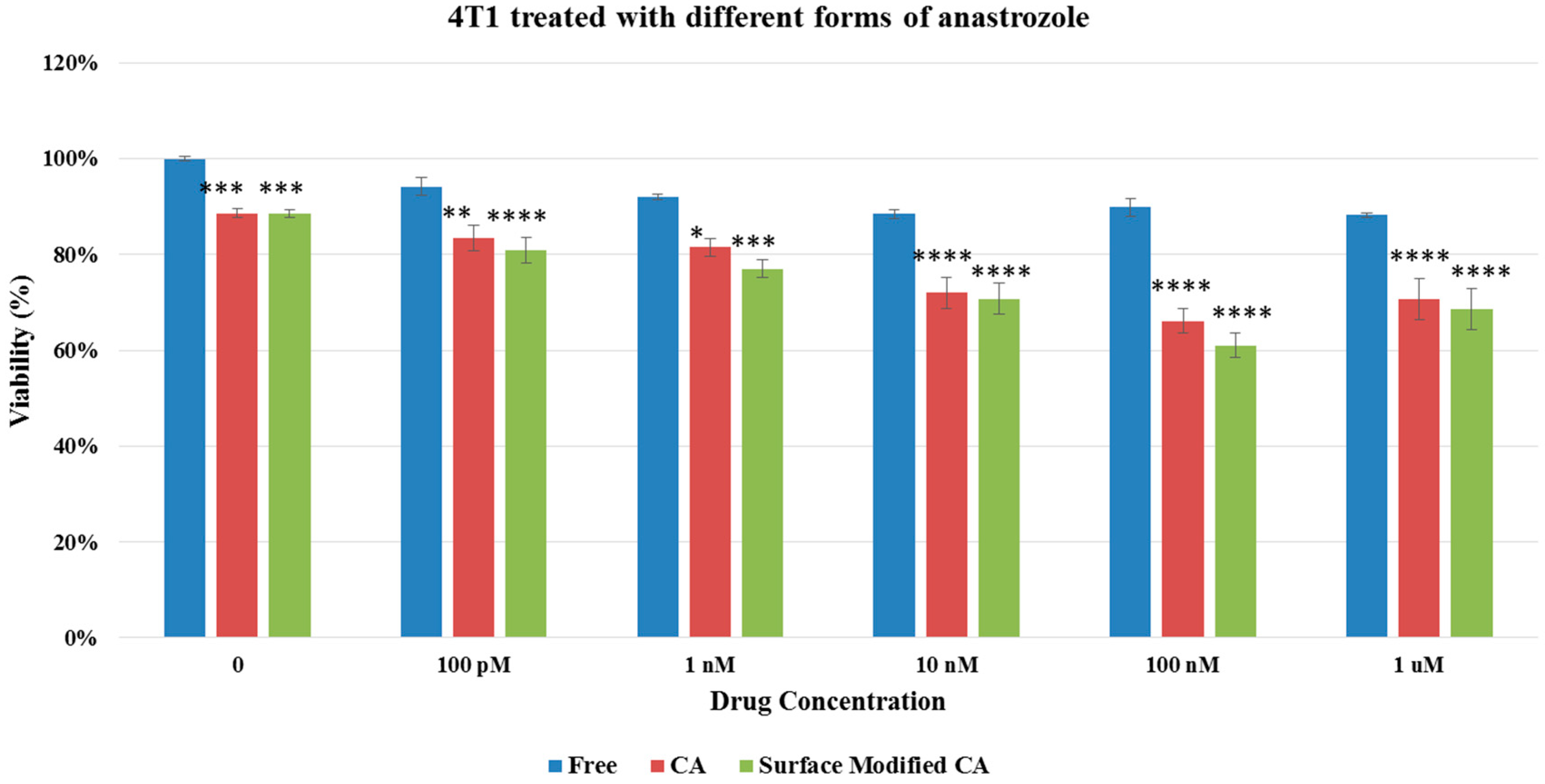
Figure 8.
Cell viability of 4T1 cells after 48 h of treatment with anastrozole-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were significant (*) at p value 0.01 to 0.05, very significant (**) at p value 0.001 to 0.01, extremely significant (***) at p value 0.0001 to 0.001, and extremely significant (****) at p value < 0.0001.
Figure 8.
Cell viability of 4T1 cells after 48 h of treatment with anastrozole-loaded CA and surface modified, gemcitabine-loaded CA. Surface-modified CA denotes the particles modified with streptavidin–biotin–PEG and fibronectin. Values were significant (*) at p value 0.01 to 0.05, very significant (**) at p value 0.001 to 0.01, extremely significant (***) at p value 0.0001 to 0.001, and extremely significant (****) at p value < 0.0001.
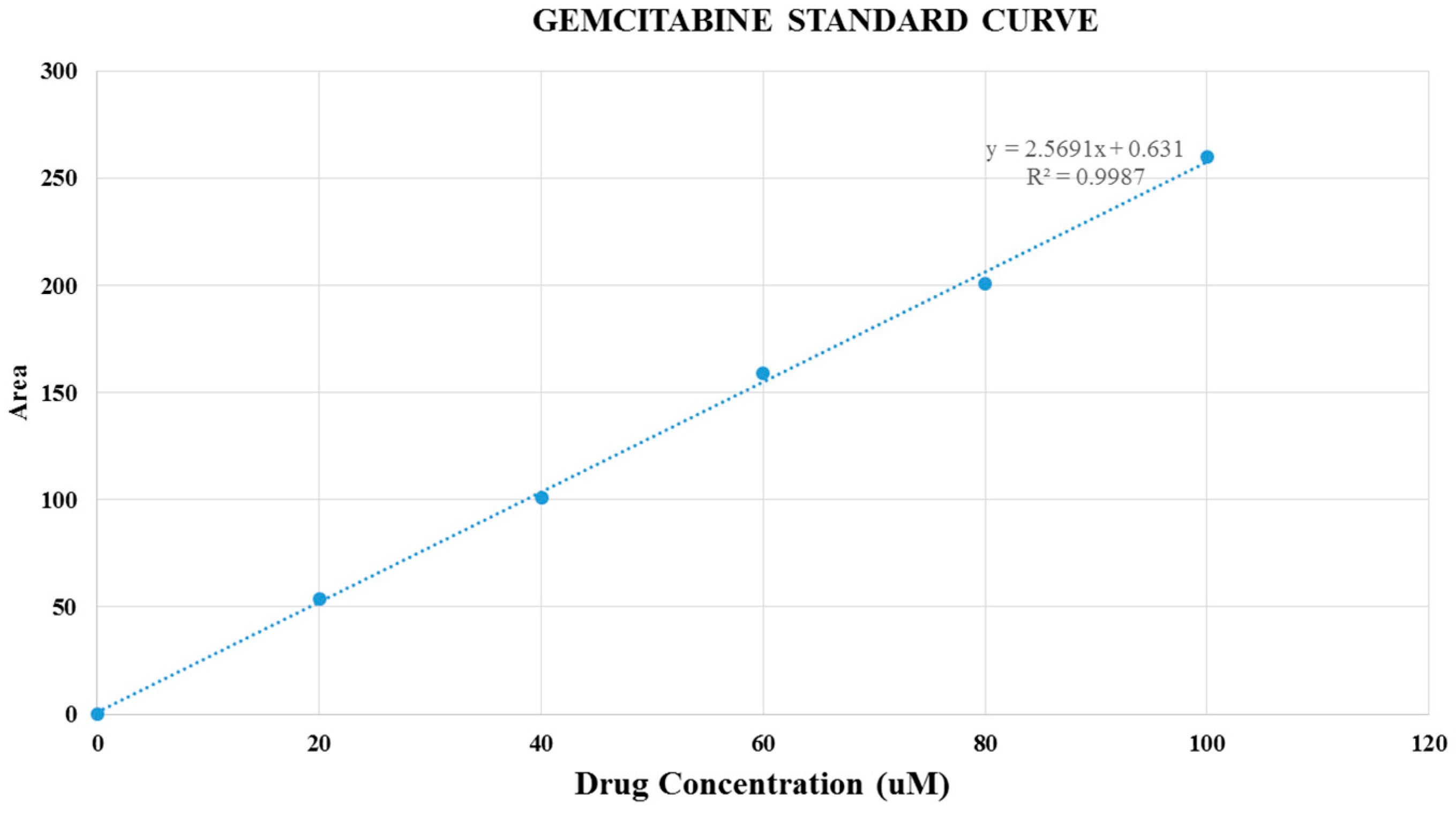
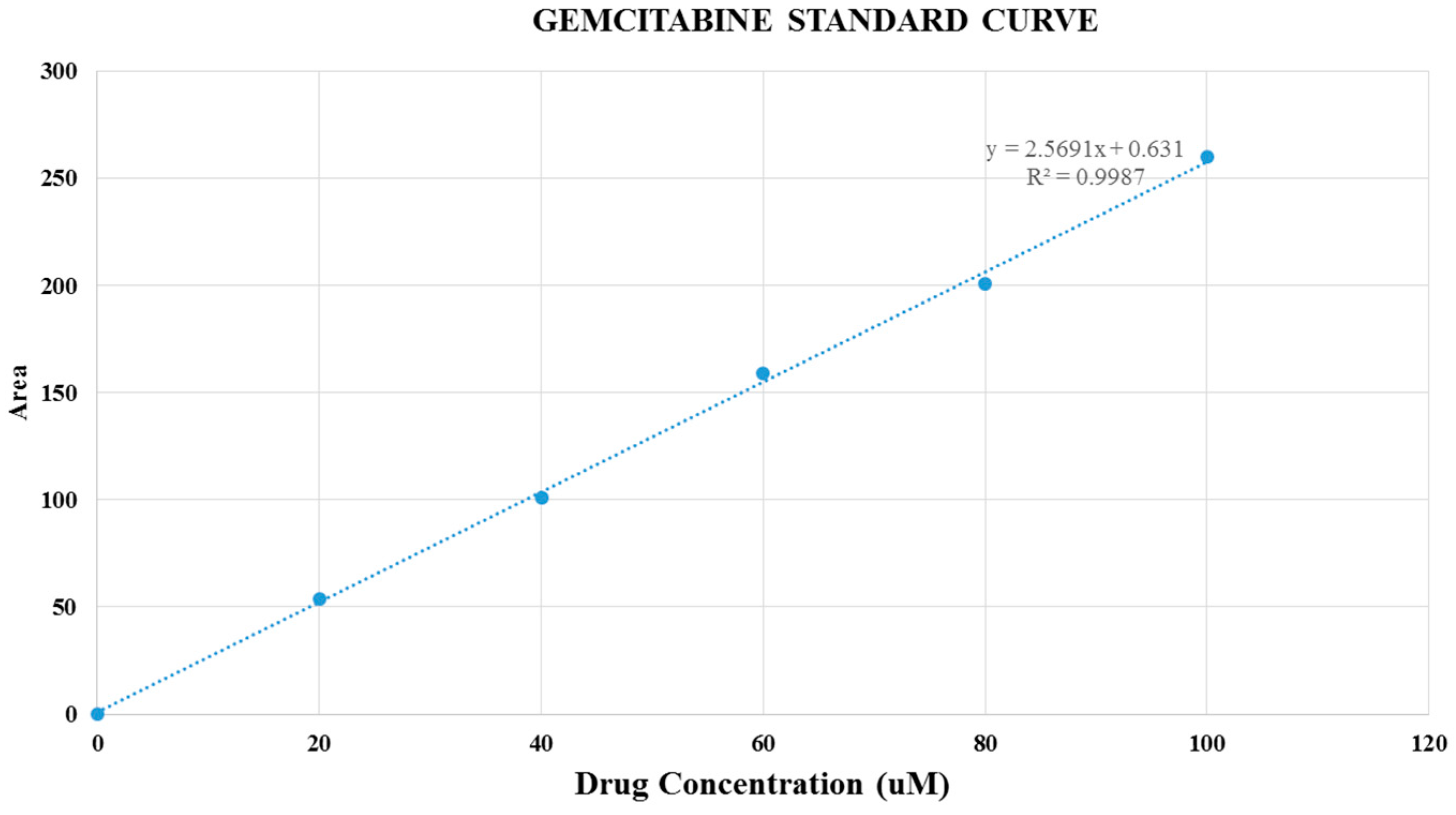
Figure 9.
Gemcitabine standard curve.
Figure 9.
Gemcitabine standard curve.
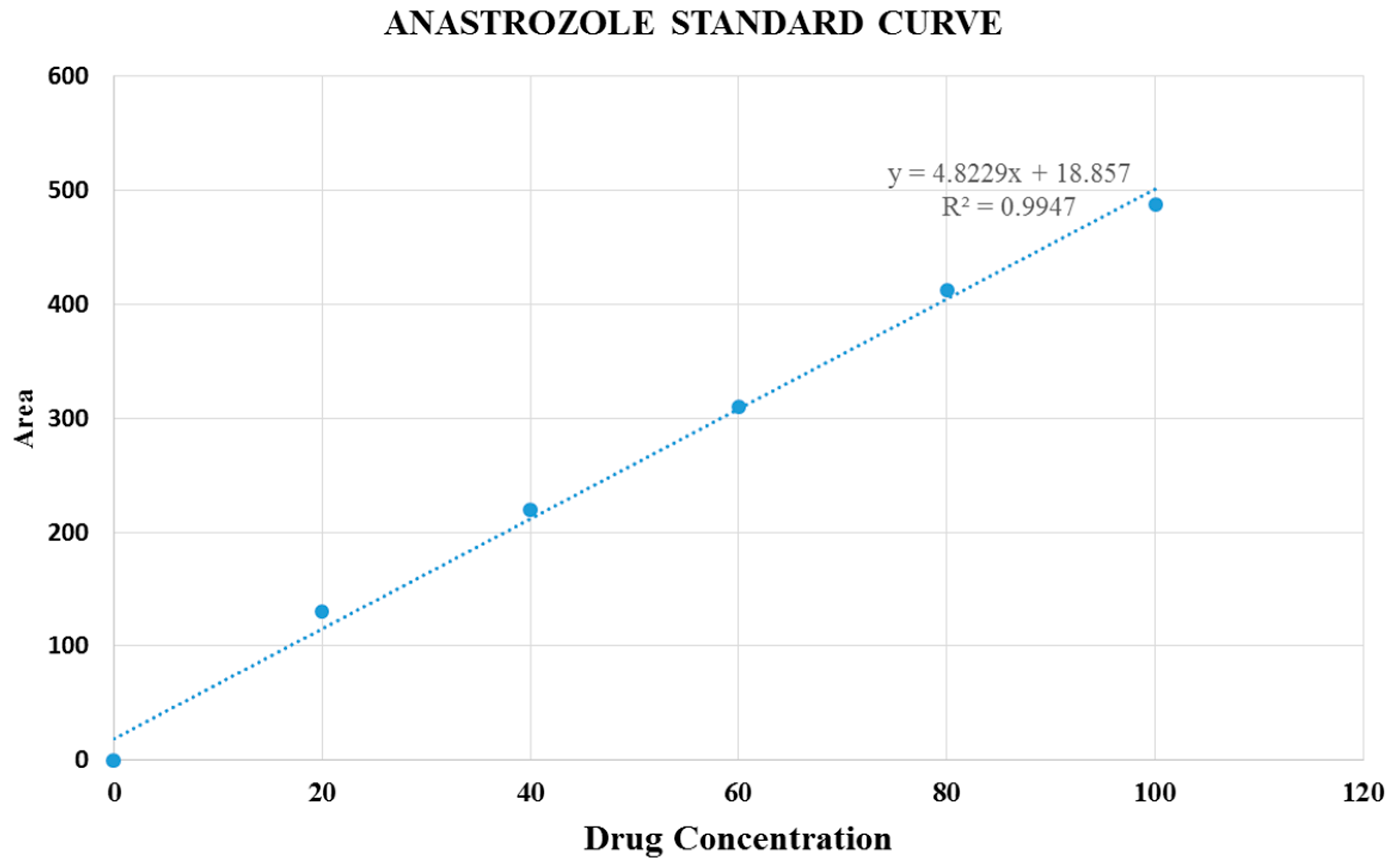
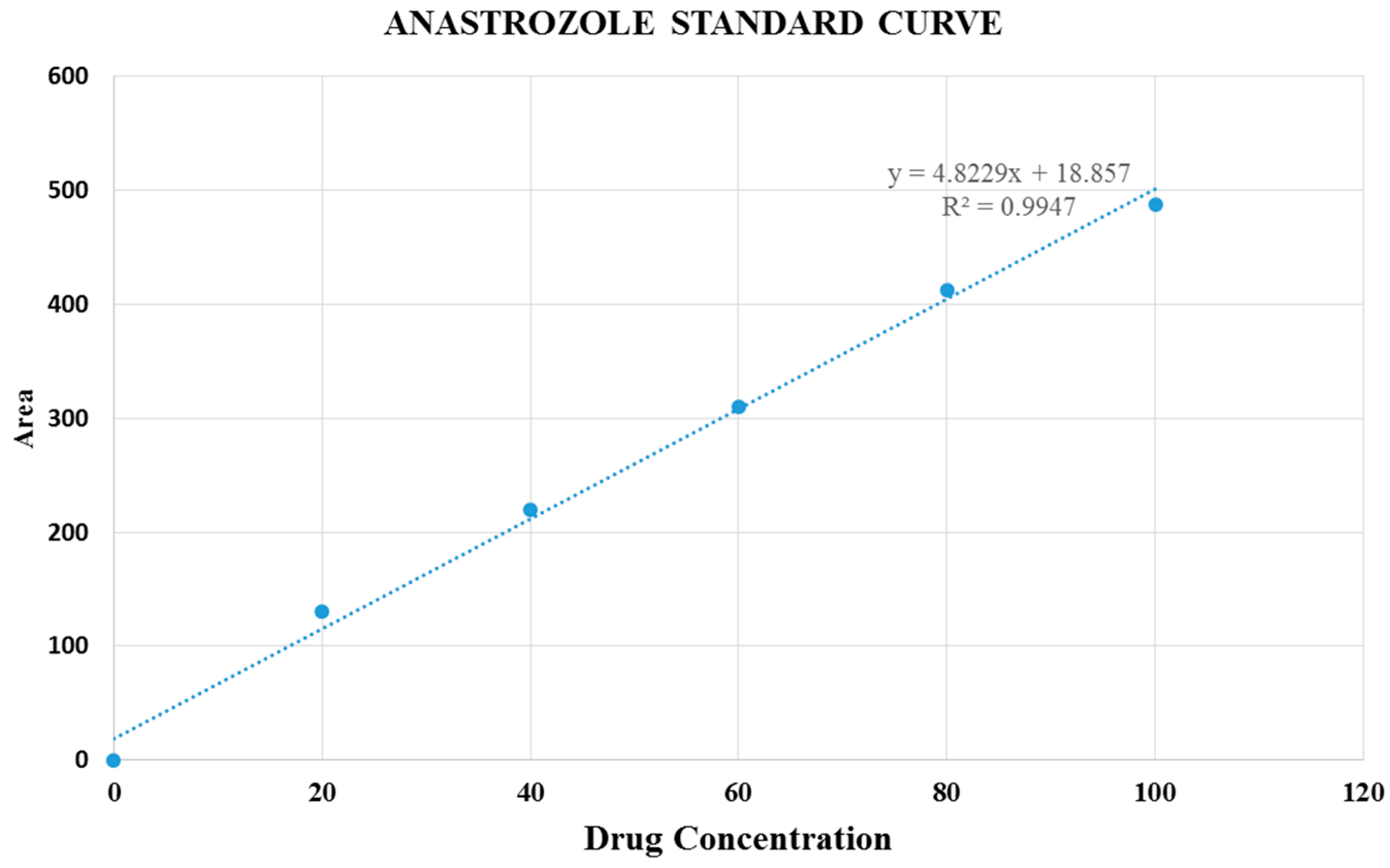
Figure 10.
Standard curve of anastrozole.
Figure 10.
Standard curve of anastrozole.
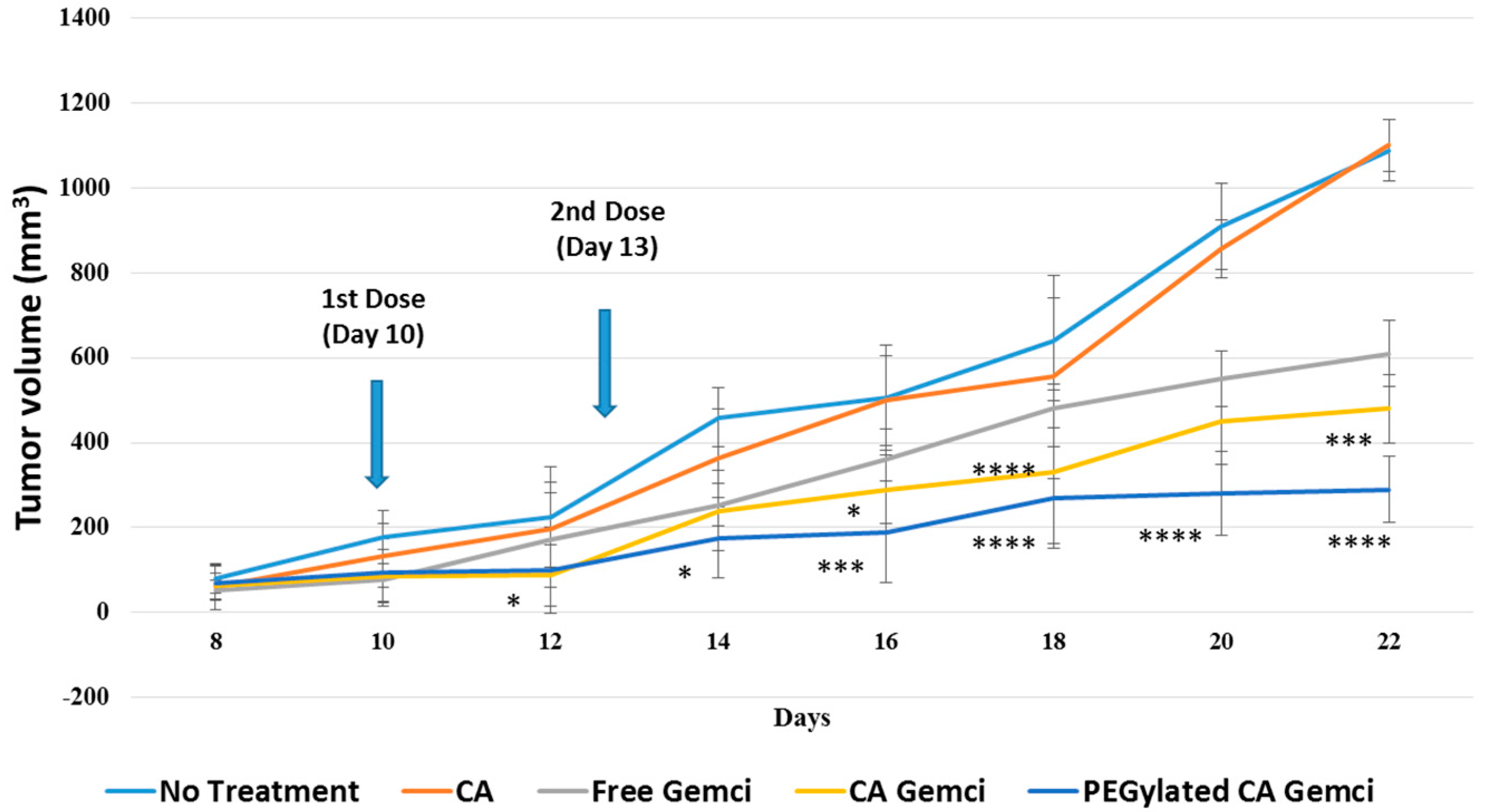
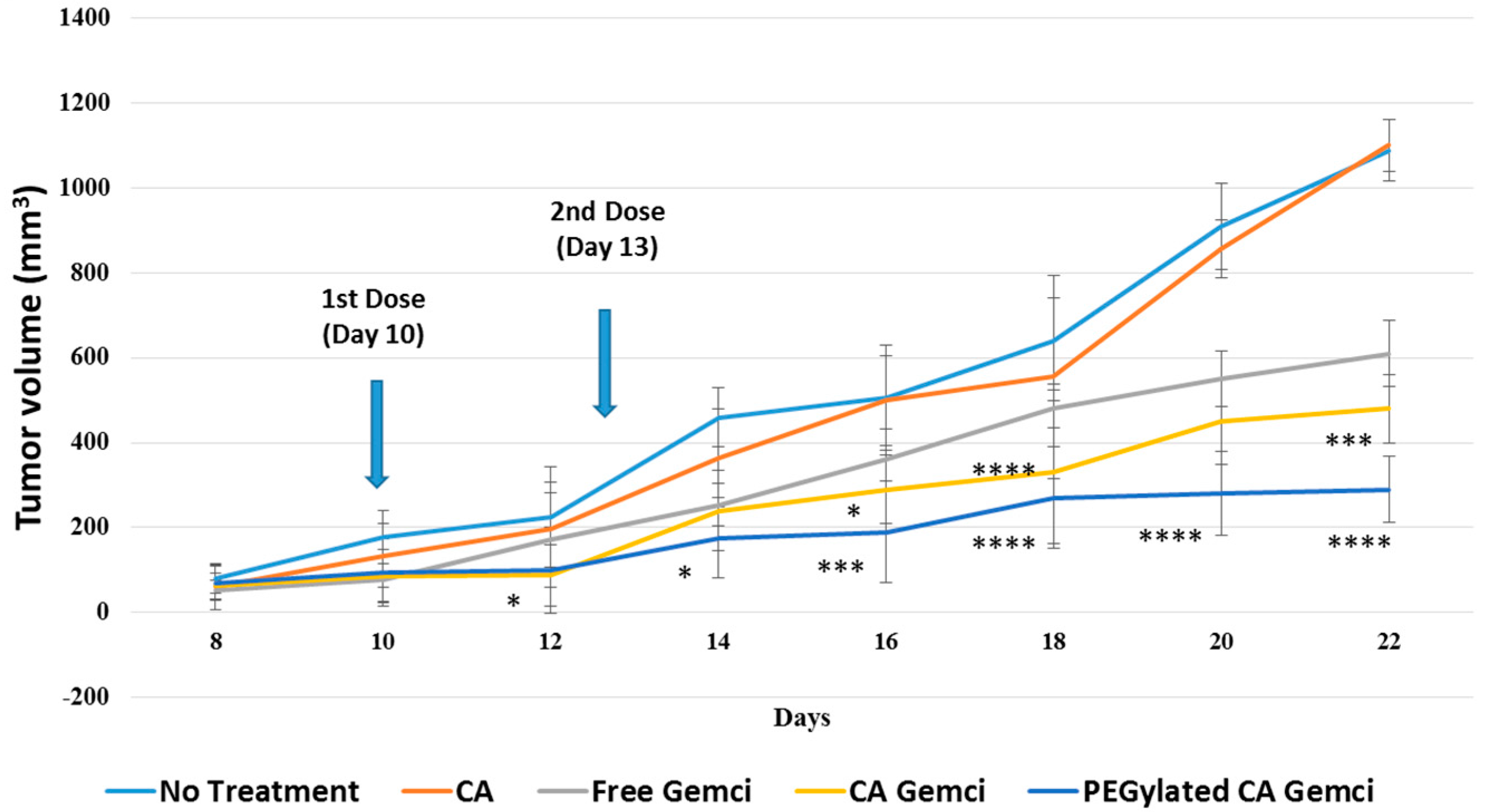
Figure 11.
Tumor regression study following intravenous delivery of gemcitabine-loaded nanoparticles to 4T1-induced breast tumors in mice. 4T1 cells were inoculated subcutaneously on the mammary pad of mice. Tumor-bearing mice were treated intravenously through tail vein injection with 100 µL of free gemcitabine (0.5 mg/kg) and surface-modified or unmodified nanoparticles (formed with 0.5 mg gemcitabine/kg), when the tumor volume reached to 13.20 ± 2.51 mm3. Six mice per group were used and data were represented as mean ± SD of tumor volume. Values were significant (*) at p value 0.01 to 0.05, very significant (**) at p value 0.001 to 0.01, extremely significant (***) at p value 0.0001 to 0.001, and extremely significant (****) at p value < 0.0001.
Figure 11.
Tumor regression study following intravenous delivery of gemcitabine-loaded nanoparticles to 4T1-induced breast tumors in mice. 4T1 cells were inoculated subcutaneously on the mammary pad of mice. Tumor-bearing mice were treated intravenously through tail vein injection with 100 µL of free gemcitabine (0.5 mg/kg) and surface-modified or unmodified nanoparticles (formed with 0.5 mg gemcitabine/kg), when the tumor volume reached to 13.20 ± 2.51 mm3. Six mice per group were used and data were represented as mean ± SD of tumor volume. Values were significant (*) at p value 0.01 to 0.05, very significant (**) at p value 0.001 to 0.01, extremely significant (***) at p value 0.0001 to 0.001, and extremely significant (****) at p value < 0.0001.
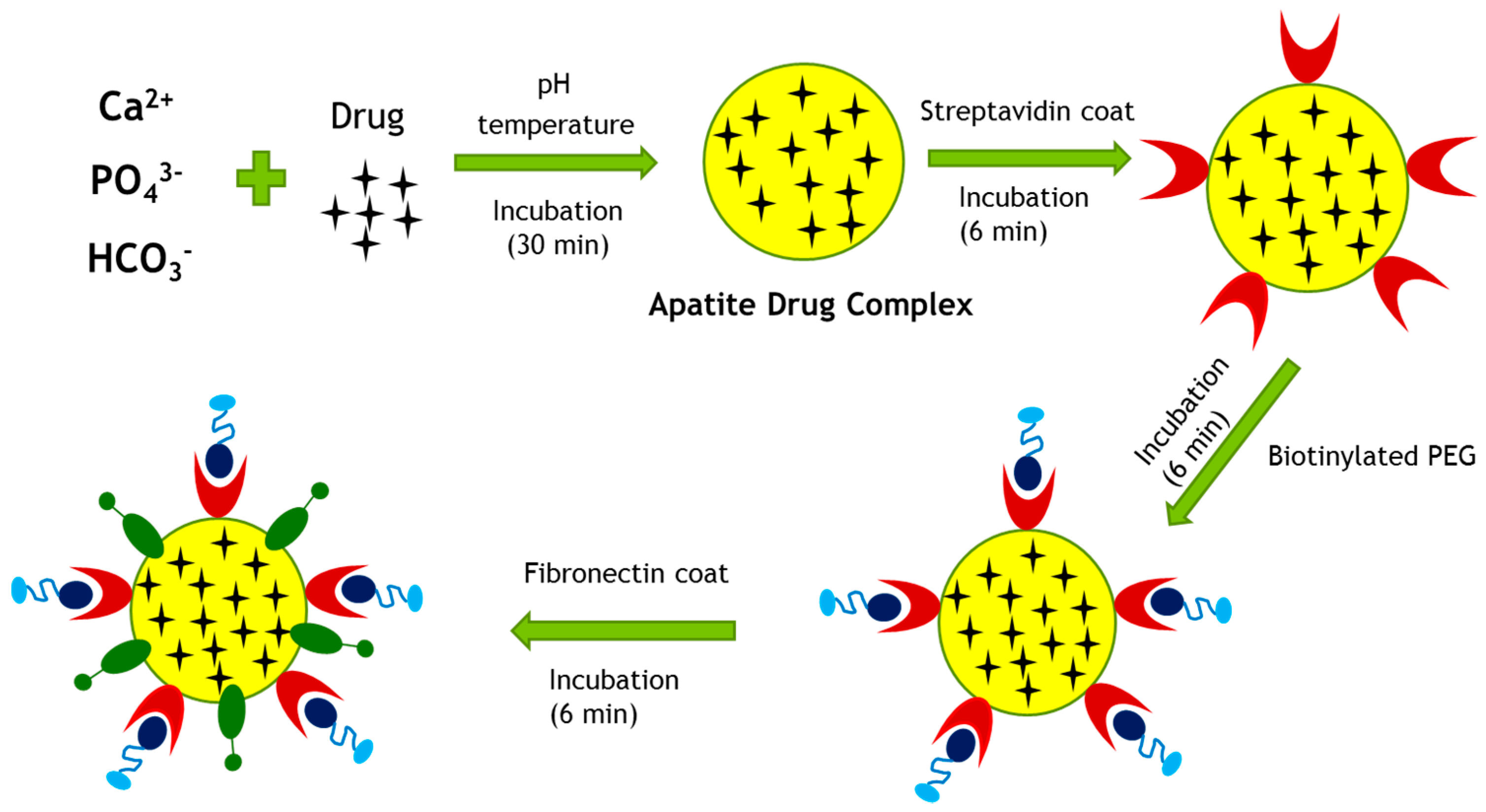
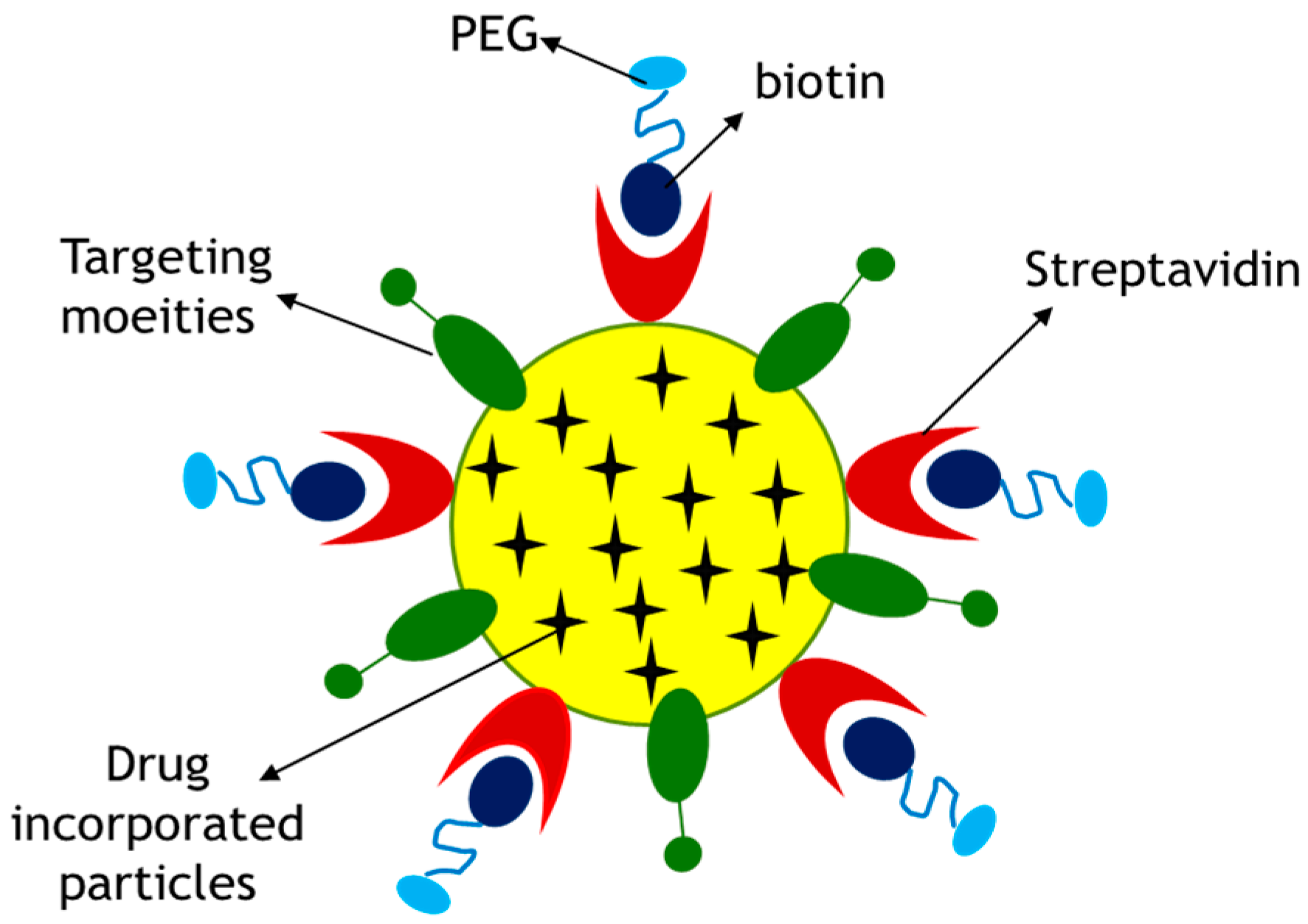
Figure 12.
Proposed surface modification model for a drug-loaded CA particle. Surface modification involves PEGylation and attachment of a targeting moiety to drug–apatite complexes. Streptavidin was used as a linker between drug–apatite particles and biotin–PEG, since biotin has specific affinity towards straptavidin. A cell-targeting moiety, such as fibronectin, is attached to the particles via ionic interactions.
Figure 12.
Proposed surface modification model for a drug-loaded CA particle. Surface modification involves PEGylation and attachment of a targeting moiety to drug–apatite complexes. Streptavidin was used as a linker between drug–apatite particles and biotin–PEG, since biotin has specific affinity towards straptavidin. A cell-targeting moiety, such as fibronectin, is attached to the particles via ionic interactions.
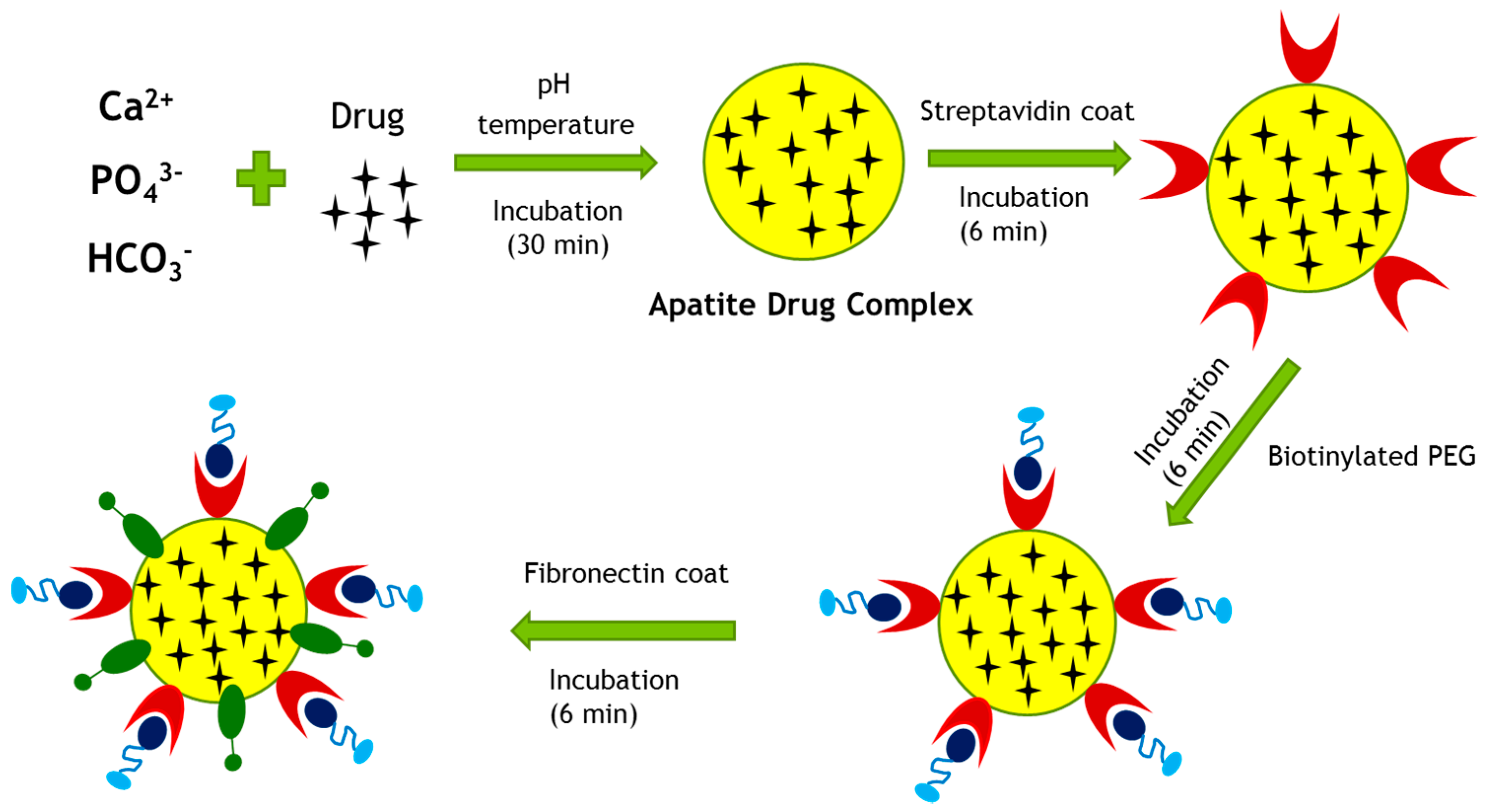
Figure 13.
Fabrication of surface-modified, drug-loaded CA particles. The particles were fabricated by allowing interactions among the salts of Ca2+, inorganic phosphate and bicarbonate in DMEM with its pH adjusted to 7.4, and incubating the resulting mixture at 37 °C for 30 min. Once the apatite–drug complex were formed, streptavidin, biotinylated PEG and fibronectin were added sequentially, with 6 min incubation at 37 °C maintained for each of the steps.
Figure 13.
Fabrication of surface-modified, drug-loaded CA particles. The particles were fabricated by allowing interactions among the salts of Ca2+, inorganic phosphate and bicarbonate in DMEM with its pH adjusted to 7.4, and incubating the resulting mixture at 37 °C for 30 min. Once the apatite–drug complex were formed, streptavidin, biotinylated PEG and fibronectin were added sequentially, with 6 min incubation at 37 °C maintained for each of the steps.
Table 1.
Experimental conditions maintained for HPLC experiments for gemcitabine.
Table 1.
Experimental conditions maintained for HPLC experiments for gemcitabine.
| Solvent | H2O |
| Mobile Phase | 70:30 ACN:H2O |
| Flow Rate | 1 mL/min |
| Column Temp | 30° C |
| Detection | DAD: 254 nm |
| Run Time | 10 min |
| Retention time | 2.2 min |
Table 2.
Experimental condition maintained for HPLC experiments for anastrozole.
Table 2.
Experimental condition maintained for HPLC experiments for anastrozole.
| Solvent | DMSO |
| Mobile Phase | 40:60 ACN:MiliQ |
| Flow Rate | 1 mL/min |
| Column Temp | 30° C |
| Detection | DAD: 215 nm |
| Run Time | 10 min |
| Retention time | 4.8 min |
Table 3.
Enhancement of cytotoxicity (%) for gemcitabine-loaded CA particles in MCF7.
Table 3.
Enhancement of cytotoxicity (%) for gemcitabine-loaded CA particles in MCF7.
| | 100 pM | 1 nM | 10 nM | 100 nM | 1 µM |
|---|
| Uncoated | 1.4 ± 1.8 | 2.8 ± 2 | 7.9 ± 1.8 | 10.8 ± 1.6 | 17.65 ± 2.7 |
| Coated | 1.9 ± 1.2 | 3.1 ± 1.1 | 9.2 ± 1.4 | 12.1 ± 1.9 | 20.4 ± 2.4 |
Table 4.
Enhancement of cytotoxicity (%) for gemcitabine-loaded CA in 4T1.
Table 4.
Enhancement of cytotoxicity (%) for gemcitabine-loaded CA in 4T1.
| | 100 pM | 1 nM | 10 nM | 100 nM | 1 µM |
|---|
| Uncoated | 1 ± 1.8 | 3.2 ± 1.2 | 3.5 ± 1.7 | 5.7 ± 2 | 10.4 ± 1.3 |
| Coated | 1.5 ± 1.9 | 3.8 ± 1.1 | 3.6 ± 1.15 | 8.5 ± 1.6 | 11.7 ± 1.5 |
Table 5.
Enhancement of cytotoxicity (%) for anastrozole-loaded CA in MCF7.
Table 5.
Enhancement of cytotoxicity (%) for anastrozole-loaded CA in MCF7.
| | 100 pM | 1 nM | 10 nM | 100 nM | 1 µM |
|---|
| Uncoated | 1.1 ± 2.5 | 1.34 ± 2 | 4.2 ± 1.4 | 5.04 ± 2.2 | 0.8 ± 1.7 |
| Coated | 1.8 ± 1.4 | 3.7 ± 2.3 | 6.1 ± 2.4 | 7.2 ± 2.3 | 3 ± 1.9 |
Table 6.
Enhancement of cytotoxicity (%) for anastrozole-loaded CA in 4T1.
Table 6.
Enhancement of cytotoxicity (%) for anastrozole-loaded CA in 4T1.
| | 100 pM | 1 nM | 10 nM | 100 nM | 1 µM |
|---|
| Uncoated | 1.6 ± 1.5 | 2 ± 2.4 | 6.3 ± 2 | 11.5 ± 2.7 | 8.7 ± 3.2 |
| Coated | 2.2 ± 2 | 4.7 ± 1.6 | 8.8 ± 2.25 | 18.4 ± 2.1 | 10.6 ± 3.76 |
Table 7.
Estimation of gemcitabine and anastrozole binding affinity to CA particles in three different drug concentrations (20, 60 and 100 μM).
Table 7.
Estimation of gemcitabine and anastrozole binding affinity to CA particles in three different drug concentrations (20, 60 and 100 μM).
| Drug Binding Affinity to CA |
|---|
| Drug | 20 µM | 60 µm | 100 µM |
| Gemcitabine | 16% ± 0.25% | 11.33% ± 0.87% | 15.16% ± 0.1% |
| Anastrozole | 0 | 0.05 ± 3 | 0 |
Table 8.
Time-dependent cellular uptake of surface-modified and unmodified, gemcitabine-loaded CA particles in MCF7 cell line. CA was formed with different Ca2+ concentrations (7, 8 and 9 mM) and 20 μM of gemcitabine. Free gemcitabine (20 μM) was used as control.
Table 8.
Time-dependent cellular uptake of surface-modified and unmodified, gemcitabine-loaded CA particles in MCF7 cell line. CA was formed with different Ca2+ concentrations (7, 8 and 9 mM) and 20 μM of gemcitabine. Free gemcitabine (20 μM) was used as control.
| 20 uM | Free Gemcitabine | CA (Ca2+ 7 mM) | Coated CA (Ca2+ 7 mM) | CA (Ca2+ 8 mM) | Coated CA (Ca2+ 8 mM) | CA (Ca2+ 9 mM) | Coated CA (Ca2+ 9 mM) |
|---|
| 1 h | 30.5% ± 1.2% | 52% ± 2% | 57% ± 1% | 55.5% ± 3.52% | 57.1% ± 1.06% | 60% ± 2% | 61.2% ± 1.3% |
| 4 h | 50.75% ± 2.3% | 79% ± 3.42% | 86.65% ± 2% | 80.8% ± 2.48% | 83.66% ± 1.6% | 81.2% ± 4.5% | 84% ± 2.4% |
| 24 h | 100% | 100% | 100% | 100% | 100% | 96,4% ± 2.5% | 95.4% ± 3.2% |
Table 9.
Time-dependent cellular uptake of surface-modified and unmodified, anastrozole-loaded CA particles in MCF7 cell line. CA was formed with different Ca2+ concentrations (7, 8 and 9 mM) and 20 μM of anastrozole. Free anastrozole (20 μM) was used as control.
Table 9.
Time-dependent cellular uptake of surface-modified and unmodified, anastrozole-loaded CA particles in MCF7 cell line. CA was formed with different Ca2+ concentrations (7, 8 and 9 mM) and 20 μM of anastrozole. Free anastrozole (20 μM) was used as control.
| 20 uM | Free Anastrozole | CA (Ca2+ 7 mM) | Coated CA (Ca2+ 7 mM) | CA (Ca2+ 8 mM) | Coated CA (Ca2+ 8 mM) | CA (Ca2+ 9 mM) | Coated CA (Ca2+ 9 mM) |
|---|
| 1 h | 18.5% ± 2.4% | 30% ± 2.4% | 51.% ± 2% | 16.5% ± 1.4% | 28.24% ± 2.7% | 11.3% ± 1.82% | 33.4% ± 2.76% |
| 4 h | 47.75% ± 2.3% | 51% ± 1.7% | 84.3% ± 1.6% | 43.2% ± 2.43% | 56.35% ± 1.8% | 31.5% ± 2.6% | 48.2% ± 1.6% |
| 24 h | 89.5% ± 3% | 93% ± 2% | 100%% | 75.4% ± 2.6% | 81% ± 1.6% | 71.42% ± 1.8% | 73.45% ± 2.2% |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}