
Post-Traumatic Cerebral Infarction: A Narrative Review of Pathophysiology, Diagnosis, and Treatment
 , , ,
, , ,
Abstract
:1. Introduction
1.1. The Health and Socioeconomic Impact of Traumatic Brain Injury
1.2. Secondary Brain Injury in Traumatic Brain Injury
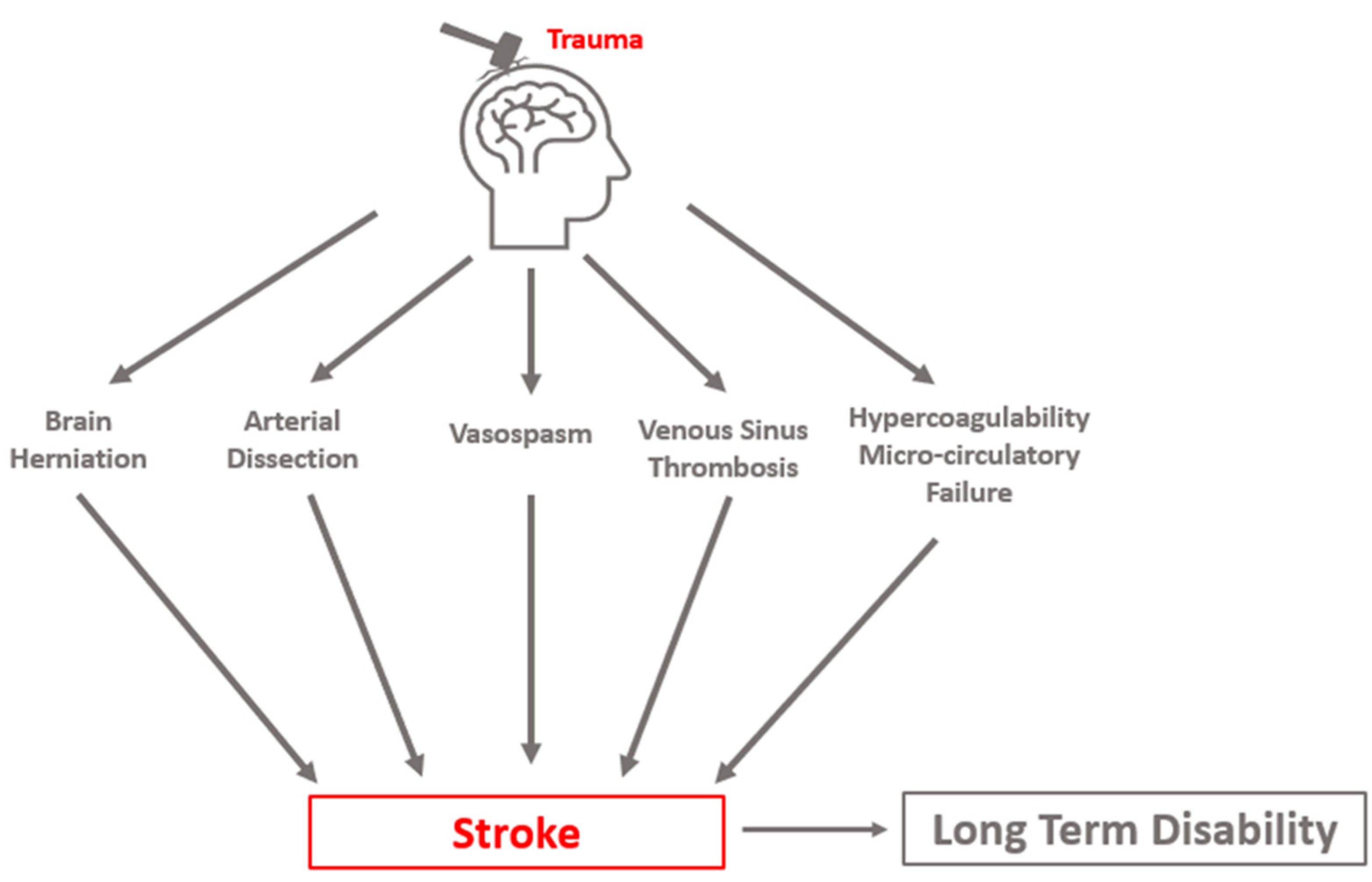
1.3. Post-Traumatic Cerebral Infarction: An Important Cause of Secondary Brain Injury
2. Pathophysiology
2.1. Hypercoagulability after Traumatic Brain Injury
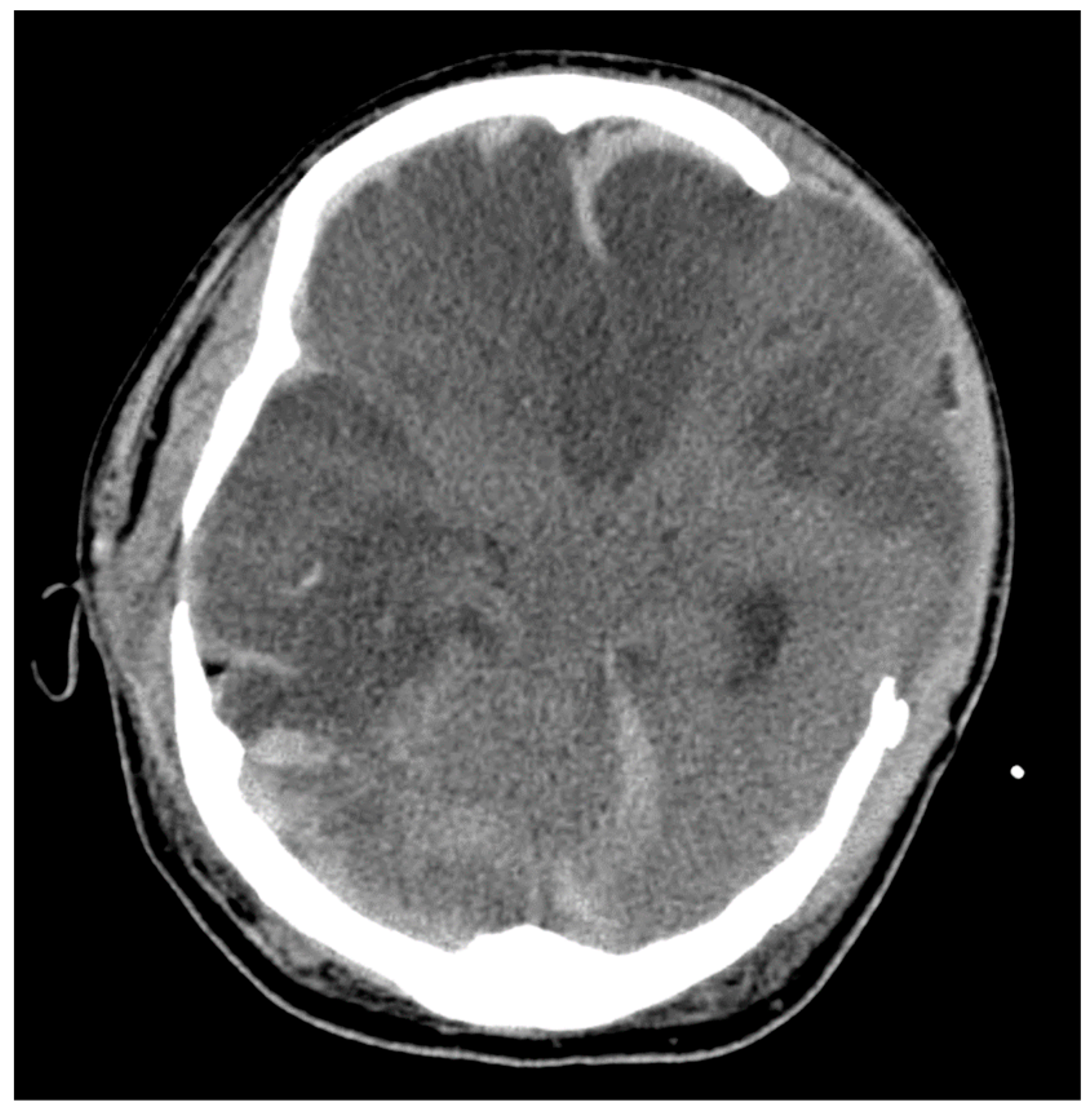
2.2. Brain Herniation
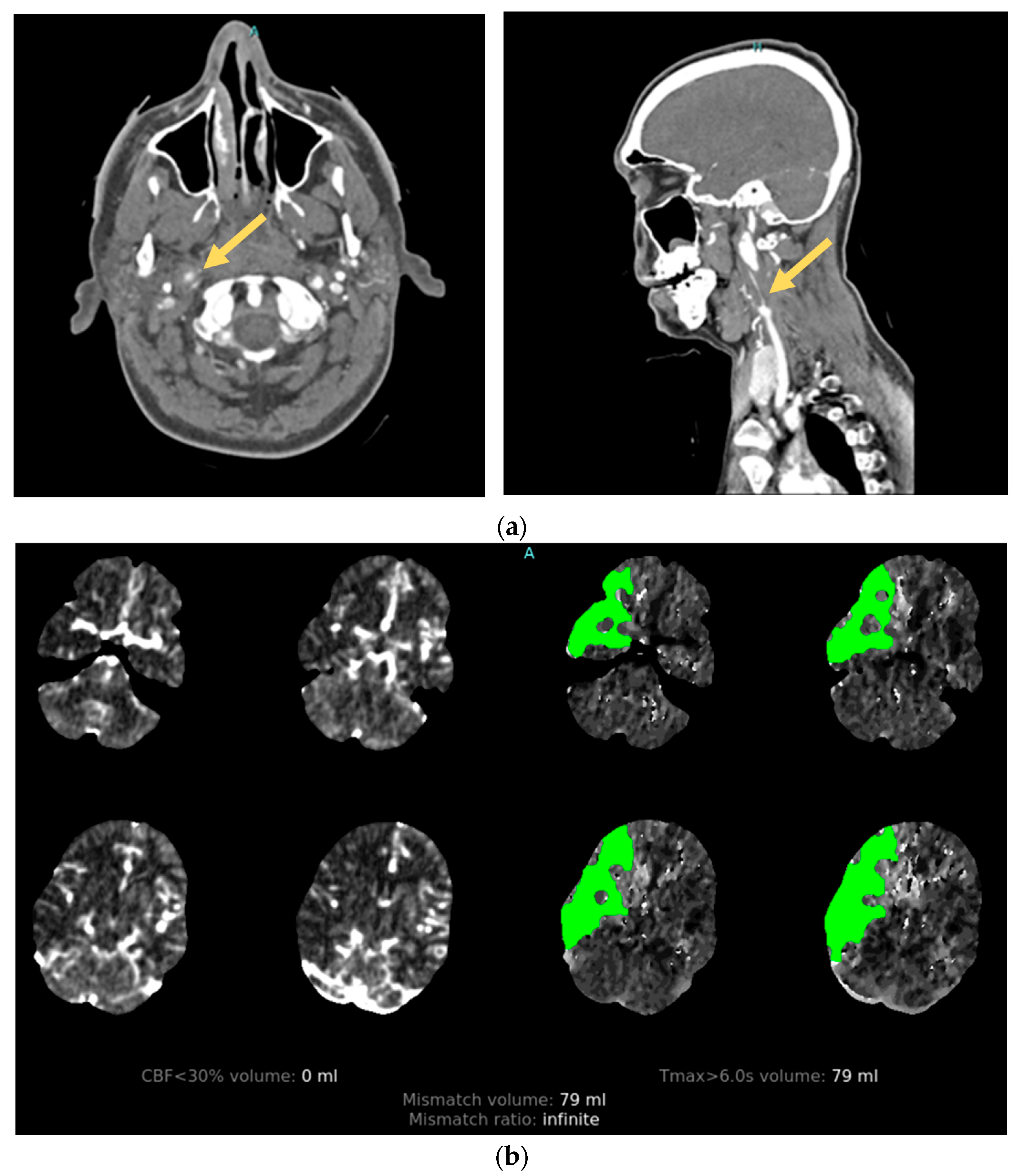
2.3. Cervical Artery Dissection
2.4. Post-Traumatic Vasospasm
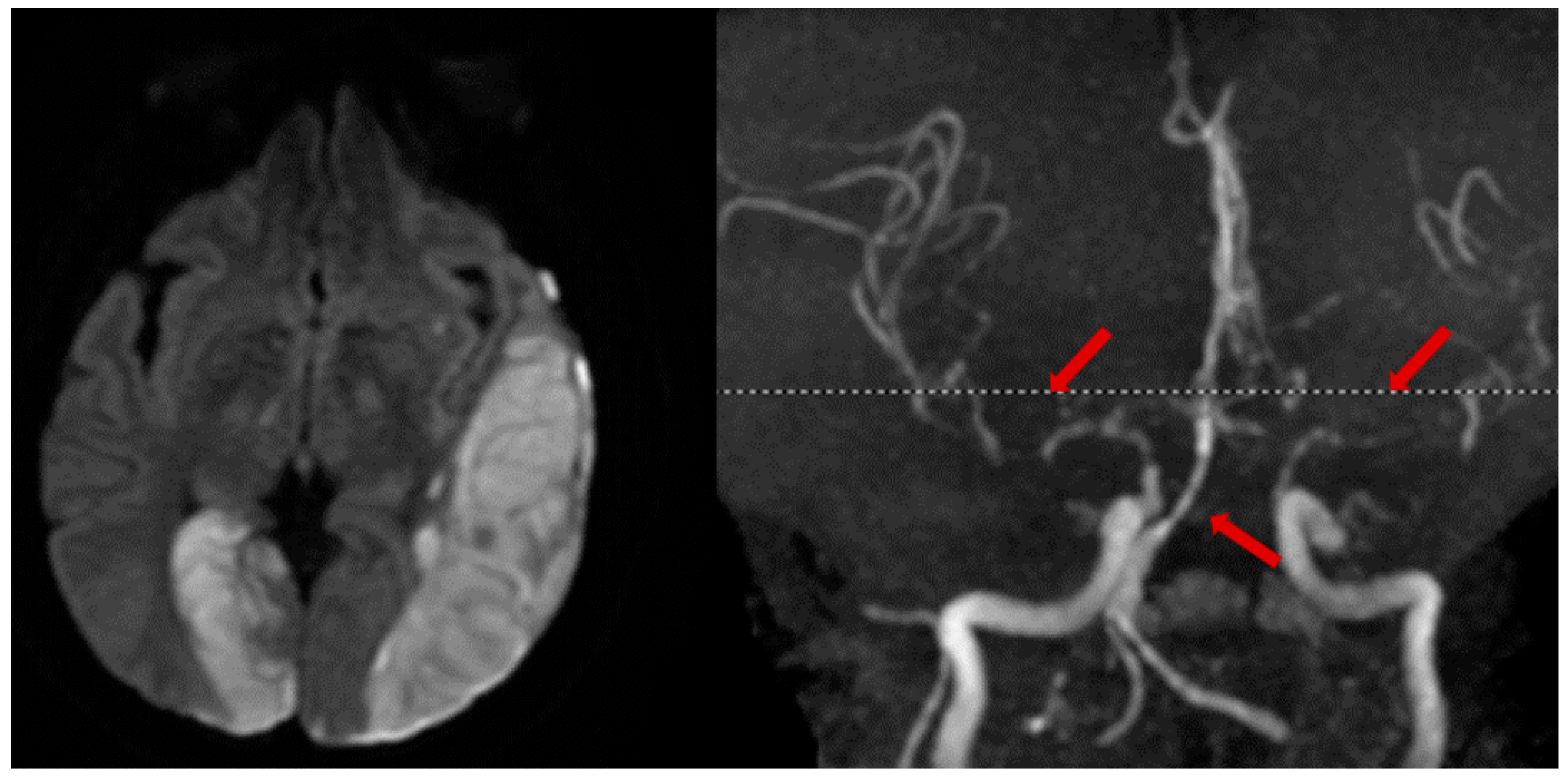
2.5. Cerebral Venous Thrombosis
2.6. Microthrombosis and Microcirculatory Failure
3. Diagnosis
3.1. Clinical Diagnosis of Acute Ischemic Stroke
3.2. CT and MRI Neuroimaging Modalities
3.3. Vessel Imaging
3.3.1. Angiography
3.3.2. MR Vessel Wall Imaging: An Emerging Diagnostic Tool
3.4. Other Methods of Detecting Ischemic Injury
3.4.1. Transcranial Doppler Ultrasound
3.4.2. Electroencephalogram
4. Management
4.1. Management of Cerebral Herniation Syndromes
4.2. Management of Cervical Artery Dissection
4.3. Management of Traumatic Vasospasm
4.4. Management of Cerebral Venous Thrombosis
5. Conclusions and Future Research Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2014; Centers for Disease Control and Prevention, U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019. [Google Scholar]
- Frieden, T.R.; Houry, D.; Baldwin, G. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation; Centers for Disease Control and Prevention, National Center for Injury Prevention and Control: Atlanta, GA, USA, 2015. [Google Scholar]
- Corso, P.; Finkelstein, E.; Miller, T.; Fiebelkorn, I.; Zaloshnja, E. Incidence and lifetime costs of injuries in the United States. Inj. Prev. 2006, 12, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Coronado, V.G.; McGuire, L.C.; Sarmiento, K.; Bell, J.; Lionbarger, M.R.; Jones, C.D.; Geller, A.I.; Khoury, N.; Xu, L. Trends in Traumatic Brain Injury in the U.S. and the public health response: 1995–2009. J. Saf. Res. 2012, 43, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Hinson, H.E.; Rowell, S.; Schreiber, M. Clinical evidence of inflammation driving secondary brain injury: A systematic review. J. Trauma Inj. Infect. Crit. Care 2015, 78, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Pilitsis, J.G.; Coplin, W.M.; O’Regan, M.H.; Wellwood, J.M.; Diaz, F.G.; Fairfax, M.R.; Michael, D.B.; Phillis, J.W. Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci. Lett. 2003, 349, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.; Schluesener, H.; Mittelbronn, M.; Seid, K.; Adjodah, D.; Wehner, H.D. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathol. 2000, 100, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Poblete, R.A.; Arenas, M.; Sanossian, N.; Freeman, W.D.; Louie, S.G. The role of bioactive lipids in attenuating the neuroinflammatory cascade in traumatic brain injury. Ann. Clin. Transl. Neurol. 2020, 7, 2524–2534. [Google Scholar] [CrossRef]
- Beaumont, A.; Marmarou, A.; Hayasaki, K.; Barzo, P.; Fatouros, P.; Corwin, F.; Marmarou, C.; Dunbar, J. The permissive nature of blood brain barrier (BBB) opening in edema formation following traumatic brain injury. Acta Neurochir. Suppl. 2000, 76, 125–129. [Google Scholar] [CrossRef]
- Kowalski, R.G.; Haarbauer-Krupa, J.K.; Bell, J.M.; Corrigan, J.D.; Hammond, F.M.; Torbey, M.T.; Hofmann, M.C.; Dams-O’Connor, K.; Miller, A.C.; Whiteneck, G.G. Acute Ischemic Stroke after Moderate to Severe Traumatic Brain Injury: Incidence and Impact on Outcome. Stroke 2017, 48, 1802–1809. [Google Scholar] [CrossRef]
- Bae, D.H.; Choi, K.S.; Yi, H.J.; Chun, H.J.; Ko, Y.; Bak, K.H. Cerebral Infarction after Traumatic Brain Injury: Incidence and Risk Factors. Korean J. Neurotrauma 2014, 10, 35–40. [Google Scholar] [CrossRef]
- Tian, H.L.; Geng, Z.; Cui, Y.H.; Hu, J.; Xu, T.; Cao, H.; Chen, S.; Chen, H. Risk factors for posttraumatic cerebral infarction in patients with moderate or severe head trauma. Neurosurg. Rev. 2008, 31, 431–437, discussion 436–437. [Google Scholar] [CrossRef] [PubMed]
- Spaite, D.W.; Hu, C.; Bobrow, B.J.; Chikani, V.; Sherrill, D.; Barnhart, B.; Gaither, J.B.; Denninghoff, K.R.; Viscusi, C.; Mullins, T.; et al. Mortality and Prehospital Blood Pressure in Patients With Major Traumatic Brain Injury: Implications for the Hypotension Threshold. JAMA Surg. 2017, 152, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Spaite, D.W.; Hu, C.; Bobrow, B.J.; Chikani, V.; Barnhart, B.; Gaither, J.B.; Denninghoff, K.R.; Adelson, P.D.; Keim, S.M.; Viscusi, C.; et al. The Effect of Combined Out-of-Hospital Hypotension and Hypoxia on Mortality in Major Traumatic Brain Injury. Ann. Emerg. Med. 2016, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Talving, P.; Benfield, R.; Hadjizacharia, P.; Inaba, K.; Chan, L.S.; Demetriades, D. Coagulopathy in severe traumatic brain injury: A prospective study. J. Trauma Inj. Infect. Crit. Care 2009, 66, 55–62, discussion 61–62. [Google Scholar] [CrossRef] [PubMed]
- Lustenberger, T.; Talving, P.; Kobayashi, L.; Inaba, K.; Lam, L.; Plurad, D.; Demetriades, D. Time course of coagulopathy in isolated severe traumatic brain injury. Injury 2010, 41, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, R.; Liu, L.; Watkins, T.; Zhang, F.; Dong, J.F. Traumatic brain injury-associated coagulopathy. J. Neurotrauma 2012, 29, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Laroche, M.; Kutcher, M.E.; Huang, M.C.; Cohen, M.J.; Manley, G.T. Coagulopathy after traumatic brain injury. Neurosurgery 2012, 70, 1334–1345. [Google Scholar] [CrossRef]
- Chestnut, R.; Ghajar, J.; Maas, A.I.R.; Marion, D.W.; Servadei, F.; Teasdale, G.M.; Unterberg, A.; von Holst, H.; Walters, B.C. Early indicators of prognosis in severe traumatic brain injury: Brain Trauma Foundation. J. Neurotrauma 2000, 57, 1–103. [Google Scholar]
- Currie, S.; Saleem, N.; Straiton, J.A.; Macmullen-Price, J.; Warren, D.J.; Craven, I.J. Imaging assessment of traumatic brain injury. Postgrad. Med. J. 2015, 92, 41–50. [Google Scholar] [CrossRef]
- Lan, Z.; Richard, S.A.; Li, Q.; Wu, C.; Zhang, Q.; Chen, R.; Yang, C. Outcomes of patients undergoing craniotomy and decompressive craniectomy for severe traumatic brain injury with brain herniation: A retrospective study. Medicine 2020, 99, e22742. [Google Scholar] [CrossRef]
- Mckee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Ropper, A.H. Herniation. Handb. Clin. Neurol. 2008, 90, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Sundstrøm, T.; Grände, P.; Juul, N.; Kock-Jensen, C.; Romner, B.; Wester, K. Management of Severe Traumatic Brain Injury Evidence, Tricks, and Pitfalls; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Bouma, G.J.; Muizelaar, J.P.; Stringer, W.A.; Choi, S.C.; Fatouros, P.; Young, H.F. Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J. Neurosurg. 1992, 77, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, R.; Valadka, A.B.; Gopinath, S.P.; Robertson, C.S. Brain tissue oxygen tension response to induced hyperoxia reduced in hypoperfused brain. J. Neurosurg. 2008, 108, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Castilla, L.; Gasco, J.; Nauta, H.J.; Okonkwo, D.O.; Robertson, C.S. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg. Focus 2008, 25, E7. [Google Scholar] [CrossRef] [PubMed]
- Debette, S.; Leys, D. Cervical-artery dissections: Predisposing factors, diagnosis, and outcome. Lancet Neurol. 2009, 8, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.R.; Bergstein, J.M.; DeBord, J.R. Diagnosis, treatment, and outcome of blunt carotid arterial injuries. Am. J. Surg. 1999, 178, 190–193. [Google Scholar] [CrossRef]
- Singh, R.R.; Barry, M.C.; Ireland, A.; Bouchier Hayes, D. Current diagnosis and management of blunt internal carotid artery injury. Eur. J. Vasc. Endovasc. Surg. 2004, 27, 577–584. [Google Scholar] [CrossRef]
- Baumgartner, R.W.; Arnold, M.; Baumgartner, I.; Mosso, M.; Gönner, F.; Studer, A.; Schroth, G.; Schuknecht, B.; Sturzenegger, M. Carotid dissection with and without ischemic events: Local symptoms and cerebral artery findings. Neurology 2001, 57, 827–832. [Google Scholar] [CrossRef]
- Morel, A.; Naggar, O.; Touzé, E.; Raymond, J.; Mas, J.; Meder, J.; Oppenheim, C. Mechanism of ischemic infarct in spontaneous cervical artery dissection. Stroke 2012, 43, 1354–1361. [Google Scholar] [CrossRef]
- O’Brien, N.F.; Reuter-Rice, K.E.; Khanna, S.; Peterson, B.M.; Quinto, K.B. Vasospasm in children with traumatic brain injury. Intensiv. Care Med. 2010, 36, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Kosty, T. Cerebral vasospasm after subarachnoid hemorrhage: An update. Crit. Care Nurs. 2005, 28, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.A.; Doberstein, C.; Zane, C.; Caron, M.J.; Thomas, K.; Becker, D.P. Posttraumatic cerebral arterial spasm: Transcranial Doppler ultrasound, cerebral blood flow, and angiographic findings. J. Neurosurg. 1992, 77, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Bederson, J.B.; Connolly, E.S., Jr.; Batjer, H.H.; Dacey, R.G.; Dion, J.E.; Diringer, M.N.; Duldner, J.E., Jr.; Harbaugh, R.E.; Patel, A.B.; Rosenwasser, R.H. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke 2009, 40, 994–1025. [Google Scholar] [CrossRef] [PubMed]
- Zubkov, A.Y.; Lewis, A.I.; Raila, F.A.; Zhang, J.; Parent, A.D. Risk factors for the development of post-traumatic cerebral vasospasm. Surg. Neurol. 2000, 53, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Alford, P.W.; Dabiri, B.E.; Goss, J.A.; Hemphill, M.A.; Brigham, M.D.; Parker, K.K. Blast-induced phenotypic switching in cerebral vasospasm. Proc. Natl. Acad. Sci. USA 2011, 108, 12705–12710. [Google Scholar] [CrossRef] [PubMed]
- Compton, J.S.; Teddy, P.J. Cerebral Arterial Vasospasm Following Severe Head Injury: A Transcranial Doppler Study. Br. J. Neurosurg. 1987, 1, 435–439. [Google Scholar] [CrossRef]
- Kramer, D.R.; Winer, J.L.; Pease, B.A.; Amar, A.P.; Mack, W.J. Cerebral vasospasm in traumatic brain injury. Neurol. Res. Int. 2013, 2013, 415813. [Google Scholar] [CrossRef]
- Provencio, J.J.; Vora, N. Subarachnoid hemorrhage and inflammation: Bench to bedside and back. Semin. Neurol. 2005, 25, 435–444. (In English) [Google Scholar] [CrossRef]
- Graves, J.; Betrus, C.; Rafols, J. Situating cerebral blood flow in the pathotrajectory of head trauma. In Cerebral Blood Flow, Metabolism, and Head Trauma: The Pathotrajectory of Traumatic Brain Injury; Kreipke, C.W., Rafols, J.A., Eds.; Springer: New York, NY, USA, 2013; pp. 29–51. [Google Scholar]
- Netteland, D.F.; Mejlaender-Evjensvold, M.; Skaga, N.O.; Sandset, E.C.; Aarhus, M.; Helseth, E. Cerebral venous thrombosis in traumatic brain injury: A cause of secondary insults and added mortality. J. Neurosurg. 2021, 134, 1912–1920. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Sahito, S.; Liaqat, J.; Chandrasekaran, P.N.; Siddiq, F. Traumatic Injury of Major Cerebral Venous Sinuses Associated with Traumatic Brain Injury or Head and Neck Trauma: Analysis of National Trauma Data Bank. J. Vasc. Interv. Neurol. 2020, 11, 27–33. [Google Scholar] [PubMed]
- Delgado Almandoz, J.E.; Kelly, H.R.; Schaefer, P.W.; Lev, M.H.; Gonzalez, R.G.; Romero, J.M. Prevalence of traumatic dural venous sinus thrombosis in high-risk acute blunt head trauma patients evaluated with multidetector CT venography. Radiology 2010, 255, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G. Cerebral venous thrombosis. Circulation 2012, 125, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, K.N.; Teramura, Y.; Hamad, O.A.; Asif, S.; Duehrkop, C.; Fromell, K.; Gustafson, E.; Hong, J.; Kozarcanin, H.; Magnusson, P.U.; et al. Dangerous liaisons: Complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol. Rev. 2016, 274, 245–269. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, W.D.; Alonso, O.; Busto, R.; Prado, R.; Dewanjee, S.; Dewanjee, M.K.; Ginsberg, M.D. Widespread hemodynamic depression and focal platelet accumulation after fluid percussion brain injury: A double-label autoradiographic study in rats. J. Cereb. Blood Flow Metab. 1996, 16, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Schwarzmaier, S.M.; Kim, S.W.; Trabold, R.; Plesnila, N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J. Neurotrauma 2010, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.C.; Chen, X.H.; Sinson, G.P.; Smith, D.H. Intravascular coagulation: A major secondary insult in nonfatal traumatic brain injury. J. Neurosurg. 2002, 97, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Hopp, S.; Albert-Weissenberger, C.; Mencl, S.; Bieber, M.; Schuhmann, M.K.; Stetter, C.; Nieswandt, B.; Schmidt, P.M.; Monoranu, C.; Alafuzoff, I.; et al. Targeting coagulation factor XII as a novel therapeutic option in brain trauma. Ann. Neurol. 2016, 79, 970–982. [Google Scholar] [CrossRef]
- Shetty, V.S.; Reis, M.N.; Aulino, J.M.; Berger, K.L.; Broder, J.; Choudhri, A.F.; Kendi, A.T.; Kessler, M.M.; Kirsch, C.F.; Luttrull, M.D.; et al. ACR Appropriateness Criteria Head Trauma. J. Am. Coll. Radiol. 2016, 13, 668–679. [Google Scholar] [CrossRef]
- Ryan, M.E.; Palasis, S.; Saigal, G.; Singer, A.D.; Karmazyn, B.; Dempsey, M.E.; Dillman, J.R.; Dory, C.E.; Garber, M.; Hayes, L.L.; et al. ACR Appropriateness Criteria head trauma--child. J. Am. Coll. Radiol. 2014, 11, 939–947. [Google Scholar] [CrossRef]
- Nakano, S.; Iseda, T.; Kawano, H.; Yoneyama, T.; Ikeda, T.; Wakisaka, S. Correlation of early CT signs in the deep middle cerebral artery territories with angiographically confirmed site of arterial occlusion. AJNR Am. J. Neuroradiol. 2001, 22, 654–659. [Google Scholar]
- Allen, L.M.; Hasso, A.N.; Handwerker, J.; Farid, H. Sequence-specific MR imaging findings that are useful in dating ischemic stroke. Radiographics 2012, 32, 1285–1297, discussion 1297–1299. [Google Scholar] [CrossRef]
- Liu, S.; Wan, X.; Wang, S.; Huang, L.; Zhu, M.; Zhang, S.; Liu, X.; Xiao, Q.; Gan, C.; Li, C.; et al. Posttraumatic cerebral infarction in severe traumatic brain injury: Characteristics, risk factors and potential mechanisms. Acta Neurochir. 2015, 157, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Wernick, S.; Wells, R.G. Sequelae of temporal lobe herniation: MR imaging. J. Comput. Assist. Tomogr. 1989, 13, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Rothfus, W.E.; Goldberg, A.L.; Tabas, J.H.; Deeb, Z.L. Callosomarginal infarction secondary to transfalcial herniation. AJNR Am. J. Neuroradiol. 1987, 8, 1073–1076. [Google Scholar] [PubMed]
- Weidauer, S.; Lanfermann, H.; Raabe, A.; Zanella, F.; Seifert, V.; Beck, J. Impairment of cerebral perfusion and infarct patterns attributable to vasospasm after aneurysmal subarachnoid hemorrhage: A prospective MRI and DSA study. Stroke 2007, 38, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Hopyan, J.J.; Gladstone, D.J.; Mallia, G.; Schiff, J.; Fox, A.J.; Symons, S.P.; Buck, B.H.; Black, S.E.; Aviv, R.I. Renal safety of CT angiography and perfusion imaging in the emergency evaluation of acute stroke. Am. J. Neuroradiol. 2008, 29, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Bodanapally, U.K.; Saksobhavivat, N.; Shanmuganathan, K.; Aarabi, B.; Roy, A.K. Arterial injuries after penetrating brain injury in civilians: Risk factors on admission head computed tomography. J. Neurosurg. 2015, 122, 219–226. [Google Scholar] [CrossRef]
- Perrein, A.; Petry, L.; Reis, A.; Baumann, A.; Mertes, P.; Audibert, G. Cerebral vasospasm after traumatic brain injury: An update. Minerva Anestesiol. 2015, 81, 1219–1228. [Google Scholar]
- Naraghi, L.; Larentzakis, A.; Chang, Y.; Duhaime, A.; Kaafarani, H.; Yeh, D.D.; King, D.R.; de Moya, M.A.; Velmahos, G.C. Is CT Angiography of the Head Useful in the Management of Traumatic Brain Injury? J. Am. Coll. Surg. 2015, 220, 1027–1031. [Google Scholar] [CrossRef]
- Yuan, X.; Cui, X.; Gu, H.; Wang, M.; Dong, Y.; Cai, S.; Feng, X.; Wang, X. Evaluating cervical artery dissections in young adults: A comparison study between high-resolution MRI and CT angiography. Int. J. Cardiovasc. Imaging 2020, 36, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shan, Y.; Guo, R.; Zheng, T.; Zhang, X.; Liu, Z.; Liu, K. Three-Dimensional High-Resolution Magnetic Resonance Imaging for the Assessment of Cervical Artery Dissection. Front. Aging Neurosci. 2022, 14, 785661. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ma, G.; Zhang, P.; Kang, Q. Isolated traumatic supraclinoid internal carotid artery dissection diagnosed by high-resolution vessel wall MRI. Br. J. Neurosurg. 2021, 37, 1801–1804. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, F.; Liu, Y.; Duan, J.; Fisher, M.; Ji, X.; Meng, R.; Zhang, H.; Fan, Z.; Yang, Q. Diagnostic performance of MR black-blood thrombus imaging for cerebral venous thrombosis in real-world clinical practice. Eur. Radiol. 2021, 32, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Hakim, A.; Kurmann, C.; Pospieszny, K.; Meinel, T.R.; Shahin, M.A.; Heldner, M.R.; Umarova, R.; Jung, S.; Arnold, M.; El-Koussy, M. Diagnostic Accuracy of High-Resolution 3D T2-SPACE in Detecting Cerebral Venous Sinus Thrombosis. AJNR Am. J. Neuroradiol. 2022, 43, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, F.; Liu, Y.; Duan, J.; Meng, R.; Chen, J.; Li, D.; Fan, Z.; Fisher, M.; Yang, Q.; et al. Predictors of successful endovascular treatment in severe cerebral venous sinus thrombosis. Ann. Clin. Transl. Neurol. 2019, 6, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Fatima, N.; Shuaib, A.; Chughtai, T.S.; Ayyad, A.; Saqqur, M. The Role of Transcranial Doppler in Traumatic Brain Injury: A Systemic Review and Meta-Analysis. Asian J. Neurosurg. 2019, 14, 626–633. [Google Scholar] [CrossRef]
- Ract, C.; Le Moigno, S.; Bruder, N.; Vigué, B. Transcranial Doppler ultrasound goal-directed therapy for the early management of severe traumatic brain injury. Intensiv. Care Med. 2007, 33, 645–651. [Google Scholar] [CrossRef]
- Tazarourte, K.; Atchabahian, A.; Tourtier, J.P.; David, J.; Ract, C.; Savary, D.; Monchi, M.; Vigué, B. Pre-hospital transcranial Doppler in severe traumatic brain injury: A pilot study. Acta Anaesthesiol. Scand. 2011, 55, 422–428. [Google Scholar] [CrossRef]
- Tsivgoulis, G.; Sharma, V.K.; Lao, A.Y.; Malkoff, M.D.; Alexandrov, A.V. Validation of transcranial Doppler with computed tomography angiography in acute cerebral ischemia. Stroke 2007, 38, 1245–1249. [Google Scholar] [CrossRef]
- Demchuk, A.M.; Christou, I.; Wein, T.H.; Felberg, R.A.; Malkoff, M.; Grotta, J.C.; Alexandrov, A.V. Accuracy and criteria for localizing arterial occlusion with transcranial Doppler. J. Neuroimaging 2000, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Zhang, X.; Tuerxun, T. EEG and SPECT changes in acute ischemic stroke. J. Neurol. Neurophysiol. 2014, 5, 2. [Google Scholar]
- Schneider, A.L.; Jordan, K.G. Regional attenuation without delta (RAWOD): A distinctive EEG pattern that can aid in the diagnosis and management of severe acute ischemic stroke. Am. J. Electroneurodiagnostic Technol. 2005, 45, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Foreman, B.; Claassen, J. Quantitative EEG for the detection of brain ischemia. Crit. Care 2012, 16, 216. [Google Scholar] [CrossRef] [PubMed]
- Kølsen-Petersen, J.A. Osmotherapy. In Management of Severe Traumatic Brain Injury: Evidence, Tricks, and Pitfalls; Sundstrom, T., Grände, P., Juul, N., Kock-Jensen, C., Romner, B., Wester, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 293–302. [Google Scholar]
- Koenig, M.A.; Bryan, M.; Lewin, J.L.; Mirski, M.A.; Geocadin, R.G.; Stevens, R.D. Reversal of transtentorial herniation with hypertonic saline. Neurology 2008, 70, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Bell, R.; Armonda, R.A.; Ling, G.S.F. Management of moderate and severe TBI. In Traumatic Brain Injury: A Clinician’s Guide to Diagnosis, Management, and Rehabilitation; Tsao, J.W., Ed.; Springer: New York, NY, USA, 2012; pp. 69–87. [Google Scholar]
- Kochanek, P.M.; Adelson, P.D.; Rosario, B.L.; Hutchison, J.; Ferguson, N.M.; Ferrazzano, P.; O’Brien, N.; Beca, J.; Sarnaik, A.; LaRovere, K.; et al. Comparison of Intracranial Pressure Measurements Before and After Hypertonic Saline or Mannitol Treatment in Children With Severe Traumatic Brain Injury. JAMA Netw. Open 2022, 5, e220891. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, G.W.J.; Rubiano, A.M.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; Shutter, L.; et al. Guidelines for the Management of Severe Traumatic Brain Injury: 2020 Update of the Decompressive Craniectomy Recommendations. Neurosurgery 2020, 87, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, P.J.; Kolias, A.G.; Timofeev, I.S.; Corteen, E.A.; Czosnyka, M.; Timothy, J.; Anderson, I.; Bulters, D.O.; Belli, A.; Eynon, C.A.; et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. N. Engl. J. Med. 2016, 375, 1119–1130. [Google Scholar] [CrossRef]
- Cooper, D.J.; Rosenfeld, J.V.; Murray, L.; Arabi, Y.M.; Davies, A.R.; D’Urso, P.; Kossman, T.; Ponsford, J.; Seppelt, I.; Reilly, P.; et al. Decompressive craniectomy in diffuse traumatic brain injury. N. Engl. J. Med. 2011, 364, 1493–1502. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.J.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2016, 80, 6–15. [Google Scholar] [CrossRef]
- Lyrer, P.; Engelter, S. Antithrombotic drugs for carotid artery dissection. Stroke 2004, 35, 613–614. [Google Scholar] [CrossRef] [PubMed]
- CADISS trial investigators. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): A randomised trial. Lancet Neurol. 2015, 14, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Wahl, W.L.; Brandt, M.M.; Thompson, B.G.; Taheri, P.A.; Greenfield, L.J. Antiplatelet therapy: An alternative to heparin for blunt carotid injury. J. Trauma Inj. Infect. Crit. Care 2002, 52, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.H.; Rahme, R.J.; Arnaout, O.; Hurley, M.C.; Berstein, R.A.; Batjer, H.H.; Bendok, B.R. Endovascular stenting of extracranial carotid and vertebral artery dissections: A systematic review of the literature. Neurosurgery 2011, 68, 856–866, discussion 866. [Google Scholar] [CrossRef]
- Vazquez Rodriguez, C.; Lemaire, V.; Renard, F.; De Keuleneer, R. Primary stenting for the acute treatment of carotid artery dissection. Eur. J. Vasc. Endovasc. Surg. 2005, 29, 350–352. [Google Scholar] [CrossRef]
- Peng, J.; Liu, Z.; Luo, C.; Chen, L.; Hou, X.; Xiao, L.; Zhou, Z. Treatment of Cervical Artery Dissection: Antithrombotics, Thrombolysis, and Endovascular Therapy. BioMed. Res. Int. 2017, 2017, 3072098. [Google Scholar] [CrossRef]
- Lee, K.H.; Lukovits, T.; Friedman, J.A. “Triple-H” therapy for cerebral vasospasm following subarachnoid hemorrhage. Neurocritical Care 2006, 4, 68–76. [Google Scholar] [CrossRef]
- Armonda, R.A.; Bell, R.S.; Vo, A.H.; Ling, G.; DeGraba, T.J.; Crandall, B.; Ecklund, J.; Campbell, W.W. Wartime traumatic cerebral vasospasm: Recent review of combat casualties. Neurosurgery 2006, 59, 1215–1225, discussion 1225. [Google Scholar] [CrossRef]
- Al-Mufti, F.; Amuluru, K.; Changa, A.; Lander, M.; Patel, N.; Wajswol, E.; Al-Marsoummi, S.; Alzubaidi, B.; Singh, I.P.; Nuoman, R.; et al. Traumatic brain injury and intracranial hemorrhage-induced cerebral vasospasm: A systematic review. Neurosurg. Focus 2017, 43, E14. [Google Scholar] [CrossRef]
- Weber, M.; Grolimund, P.; Seiler, R.W. Evaluation of posttraumatic cerebral blood flow velocities by transcranial Doppler ultrasonography. Neurosurgery 1990, 27, 106–112. [Google Scholar] [CrossRef]
- Saposnik, G.; Barinagarrementeria, F.; Brown, R.D., Jr.; Bushnell, C.D.; Cucchiara, B.; Cushman, M.; deVeber, G.; Ferro, J.M.; Tsai, F.Y. Diagnosis and management of cerebral venous thrombosis: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011, 42, 1158–1192. [Google Scholar] [CrossRef] [PubMed]
- Grangeon, L.; Gilard, V.; Ozkul-Wermester, O.; Lefaucheur, R.; Curey, S.; Gerardin, E.; Derrey, S.; Maltete, D.; Magne, N.; Triquenot, A. Management and outcome of cerebral venous thrombosis after head trauma: A case series. Rev. Neurol. 2017, 173, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Mutch, C.A.; Talbott, J.F.; Gean, A. Imaging Evaluation of Acute Traumatic Brain Injury. Neurosurg. Clin. N. Am. 2016, 27, 409–439. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnostic Modality | Advantages | Limitations |
|---|---|---|
| Physical Exam | Allows serial monitoring, no additional cost | Confounded by medication effect and distracting injuries, accuracy dependent on provider experience |
| Non-contrast CT | Rapid acquisition time | Less sensitive for early and subtle ischemic changes, requires hospital transport, radiation exposure |
| Non-contrast MRI | Increased sensitivity for early and subtle ischemic changes | Prolonged acquisition time, requires hospital transport, contraindicated with certain medical devices or foreign objects |
| CT angiography | High-resolution vessel imaging, non-invasive, rapid acquisition time | Contrast may be contraindicated in allergy or acute renal injury |
| MR angiography | Can be performed with or without contrast enhancement, non-invasive | Prolonged acquisition time, less accurate in low-flow vessels, including small and distal vessels |
| DSA | High-resolution, real-time vessel imaging | Invasive procedure, not available in all centers |
| MR VWI | High-resolution vessel imaging, non-invasive | Prolonged acquisition time, not widely available |
| TCD | Non-invasive study, performed at bedside | Low specificity and sensitivity in diagnosing PTCI |
| EEG | Non-invasive study, can be monitored continuously, performed at bedside | Low specificity and sensitivity in diagnosing PTCI, confounded by medication effect and metabolic encephalopathies |
| Disease Mechanism | Timing of PTCI | Diagnostic Modality | Treatment Options |
|---|---|---|---|
| Herniation | Delayed | CT, MRI | Hyperosmolar therapy Mannitol 0.25–1.0 g/kg Hypertonic saline bolus Surgical decompression |
| Arterial Dissection | Early | CTA, MRA DSA MR VWI | Antithrombotic therapy: Antiplatelet drugs Anticoagulation * Endovascular/surgical options |
| Vasospasm | Delayed | CTA, MRA DSA | Avoidance of hypovolemia Induced hypertension Oral nimodipine Intra-arterial treatment: Calcium-channel blocker Vasodilator Angioplasty |
| Venous Thrombosis | Early or delayed | CTV, MRV MR VWI | Conservative management Anticoagulation * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poblete, R.A.; Zhong, C.; Patel, A.; Kuo, G.; Sun, P.Y.; Xiao, J.; Fan, Z.; Sanossian, N.; Towfighi, A.; Lyden, P.D. Post-Traumatic Cerebral Infarction: A Narrative Review of Pathophysiology, Diagnosis, and Treatment. Neurol. Int. 2024, 16, 95-112. https://doi.org/10.3390/neurolint16010006
Poblete RA, Zhong C, Patel A, Kuo G, Sun PY, Xiao J, Fan Z, Sanossian N, Towfighi A, Lyden PD. Post-Traumatic Cerebral Infarction: A Narrative Review of Pathophysiology, Diagnosis, and Treatment. Neurology International. 2024; 16(1):95-112. https://doi.org/10.3390/neurolint16010006
Chicago/Turabian StylePoblete, Roy A., Charlotte Zhong, Anish Patel, Grace Kuo, Philip Y. Sun, Jiayu Xiao, Zhaoyang Fan, Nerses Sanossian, Amytis Towfighi, and Patrick D. Lyden. 2024. "Post-Traumatic Cerebral Infarction: A Narrative Review of Pathophysiology, Diagnosis, and Treatment" Neurology International 16, no. 1: 95-112. https://doi.org/10.3390/neurolint16010006