2. Case Presentation
A 36-year-old Greek woman diagnosed with transfusion-dependent β-thalassemia (TDT) (IVSI-n1/-44bp del) presented with a sudden onset of lower extremity motor impairment, pelvic paresthesia, and bladder dysfunction. She had been experiencing persistent lower back pain for the past six months, with recent aggravation over the last month, accompanied by bilateral lower limb weakness. The patient was following a systematic transfusion schedule with hemoglobin levels maintained above 9.5 g/dL every 15 days, as per guidelines. Additionally, she underwent iron chelation therapy with deferoxamine/deferiprone, but with poor compliance. Her mean hemoglobin (Hb) was 10 g/dL, and the mean ferritin level was 8500 μg/L, reaching a maximum of 12,200 μg/L (the patient’s main hematological and biochemical parameters are summarized in
Table 1). Myocardial T2* value indicated 5 milliseconds (severe myocardial iron overload), while the liver T2* value was estimated at 1 millisecond, indicative of a high iron concentration in the liver (20 mg/g dry tissue). Upon admission, vital signs were stable, with a body temperature of 37 °C, blood pressure at 110/70 mm Hg, respiratory rate of 16/min, heart rate of 90 beats per minute, and oxygen saturation at 99% on room air (FiO
2: 21%). A physical examination revealed hepatosplenomegaly and a systolic heart murmur. Neurological examination at admission demonstrated that the patient had difficulty rising from a sitting position and walking due to inability in flexing and extending the plantar fascia in both lower limbs (grade of muscle strength 2/5 in both lower limbs), along with impaired Achilles tendon reflexes. The patient also exhibited complete urinary retention and sensory cauda equina syndrome at the S3 level. The basic laboratory work-up yielded no significant findings, and the absence of acute infection was confirmed through negative results in multiple blood and urine cultures, inflammatory markers, and serological markers.
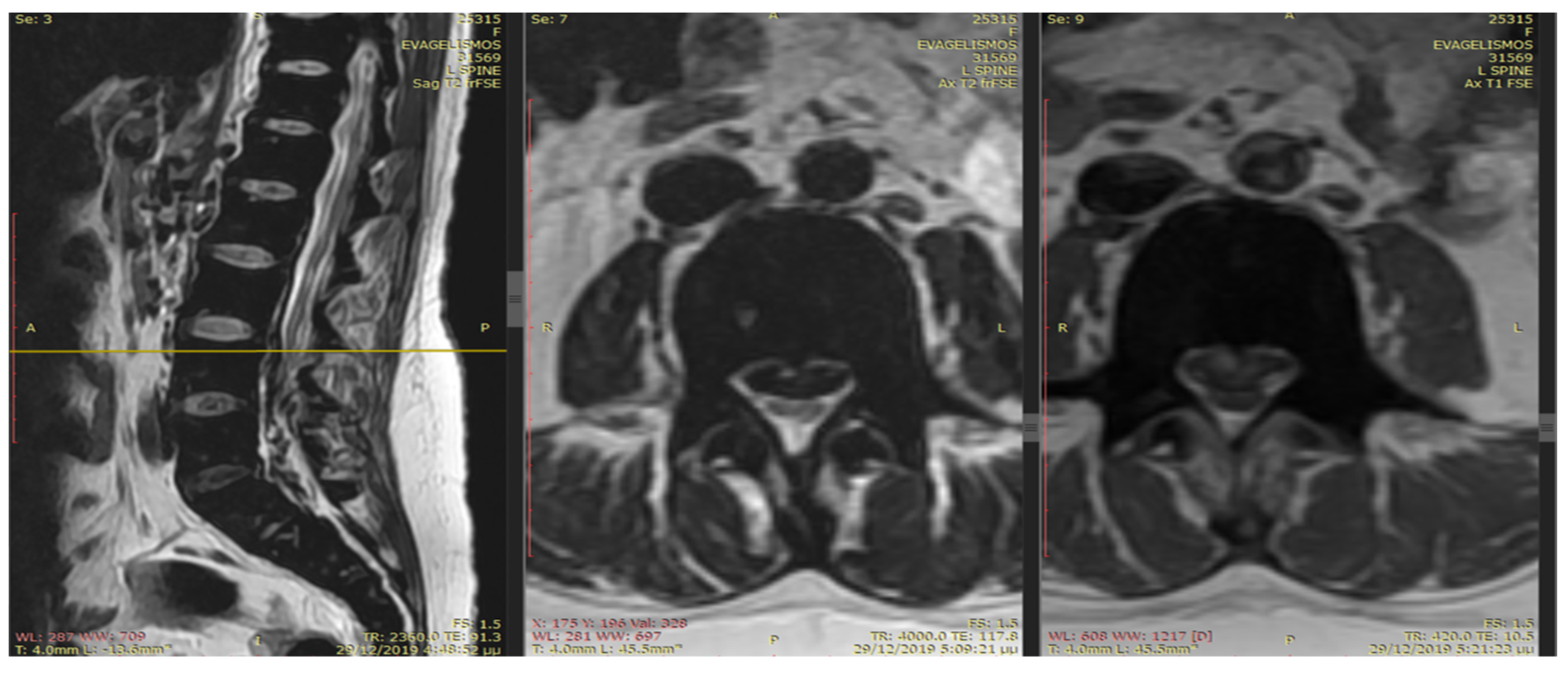
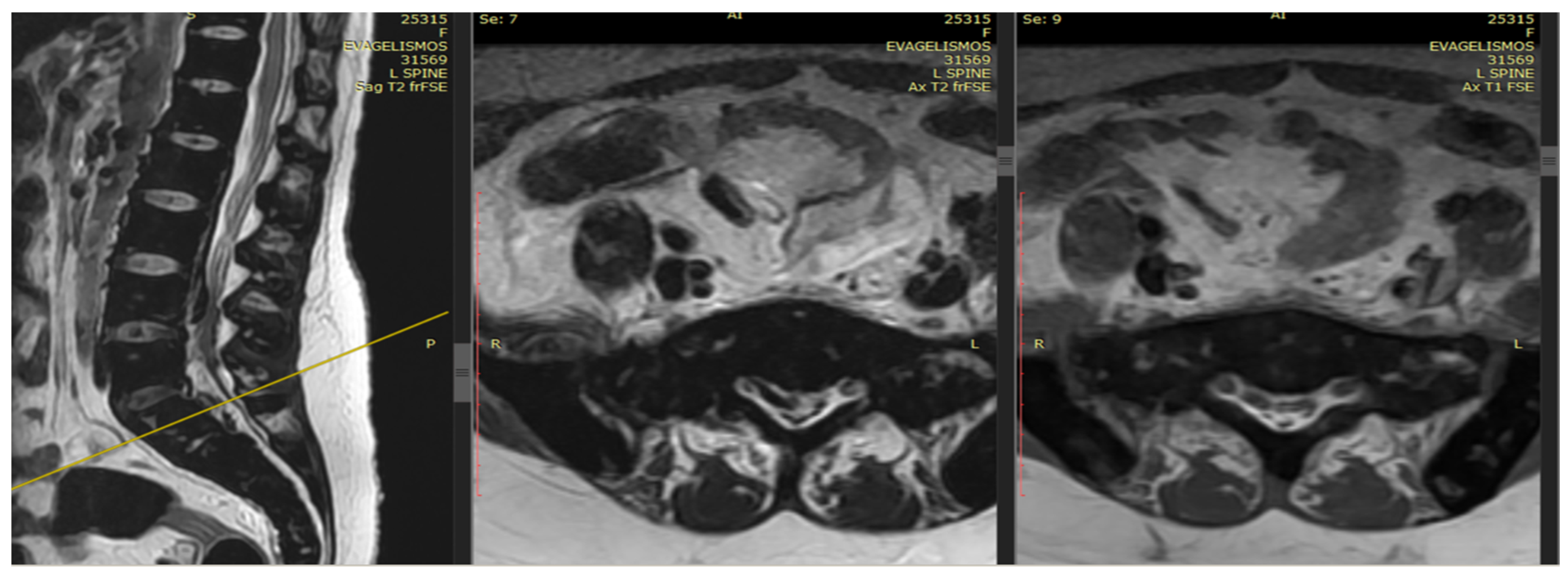
An emergency MRI was conducted on the patient, and the findings can be succinctly summarized as follows. In the T2 sequence, a low signal is observed in the anterior epidural space, accompanied by an uneven signal in the T1 sequence. These observations indicate compressive effects on the dura at the level of L2, extending towards the posterior segments of the spinal canal up to the final segment. Furthermore, a portion of this compression appears to extend along the midline fissures on the right side, particularly at the levels of L5–S1 and S1–S2, causing mild compressive effects on the nerve roots. In the GRE sequence, a low signal is noted in a segment of the aforementioned distribution, raising strong suspicion of hemosiderin deposition (
Figure 1 and
Figure 2). In the fat-suppressed T2 sequence, there is a diffuse increase in signal, signifying edema, in the soft tissues at the level of the midline fissures in the posterior elements and the soft tissues between the ligamentum flavum and the dura at the intervertebral foramina level. Additionally, edema is observed in the epidural space.
Due to the duration of symptoms exceeding 24 h, a surgical approach was ruled out as it could not be classified as an emergency. Local radiation was proposed to the patient; however, she declined the option due to plans for an upcoming pregnancy. Throughout her hospitalization, the patient underwent hypertransfusion protocols to maintain hemoglobin levels at 12 g/dL. A 24 h intravenous iron chelation regimen with deferoxamine was administered at a dosage of 100 mg/kg, while the deferiprone dosage remained constant at 70 mg/kg. Hydroxyurea was initiated at 20 mg/kg following approval.
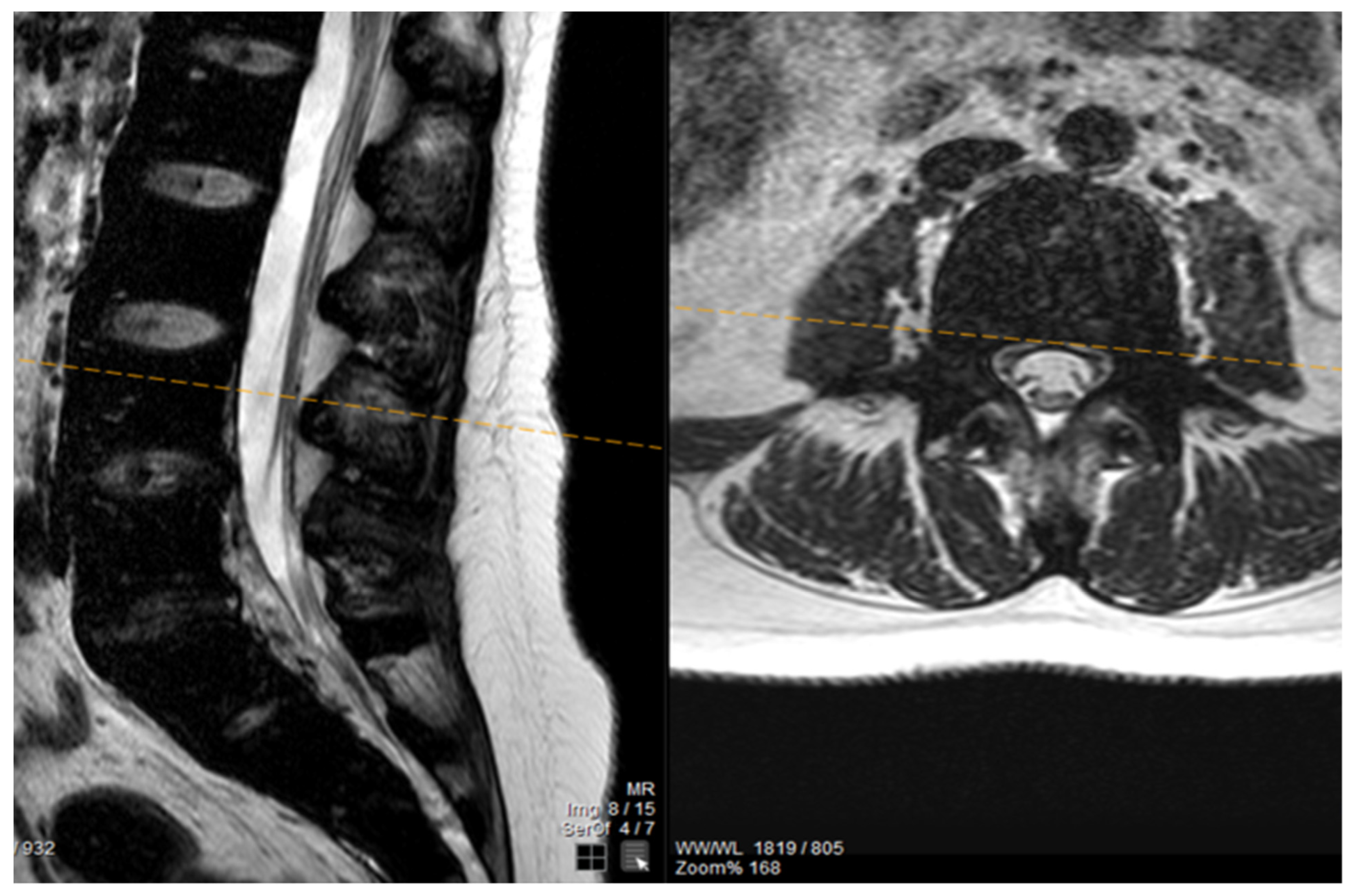
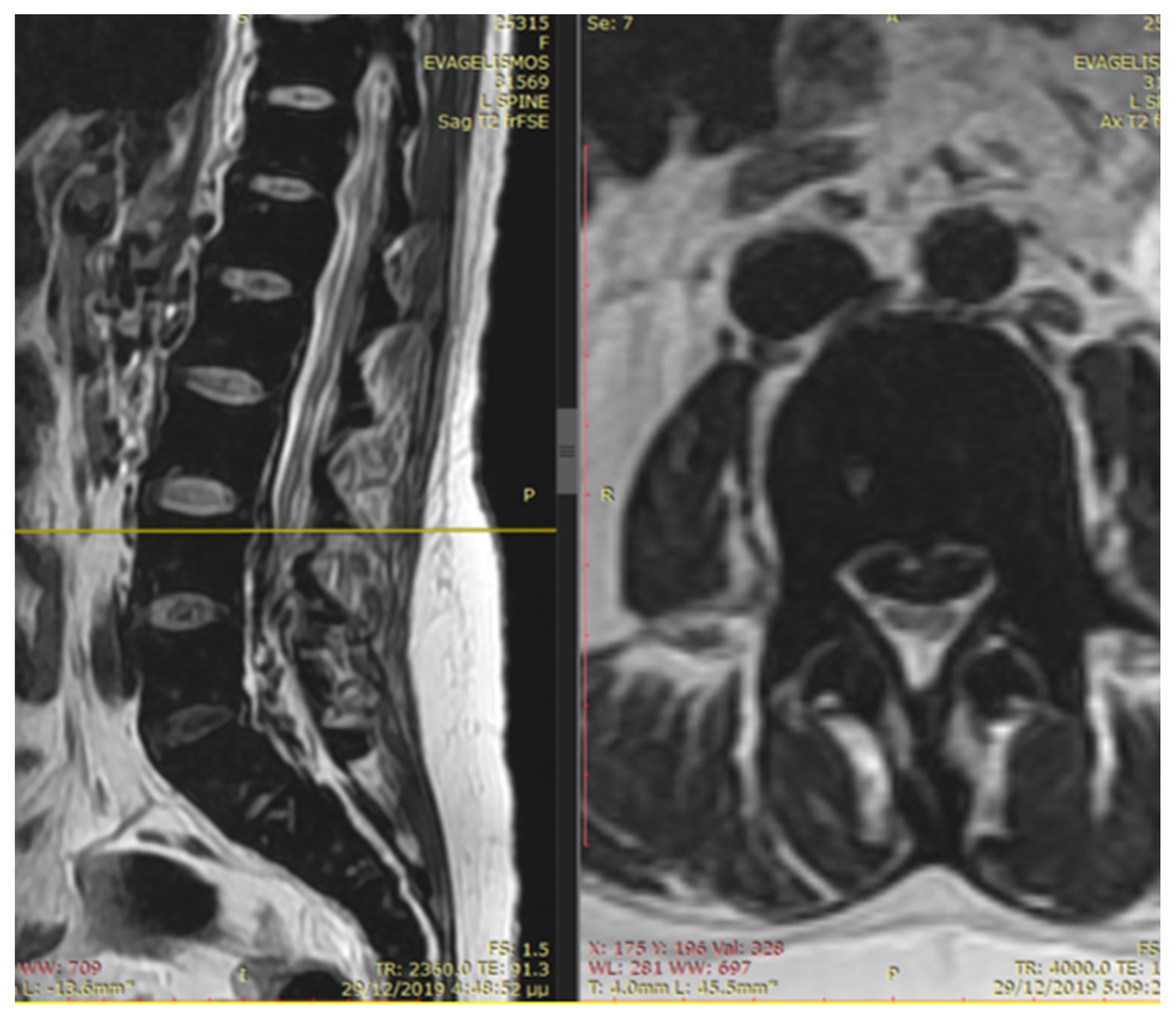
The patient noted a gradual improvement in neurological manifestations related to mobility and sensitivity. Upon discharge, she continued self-catheterization due to incomplete restoration of urination. Subsequently, the patient maintained subcutaneous iron chelation treatment with deferoxamine at a dosage of 80 mg/kg. Severe aplasia occurred two months after initiating hydroxyurea, leading to the discontinuation of both deferiprone and hydroxyurea until complete hematological recovery. Despite efforts to reintroduce hydroxyurea, the patient experienced aplasia again, prompting the decision to permanently cease this medication. Six months after the initial symptom onset, MRI findings indicated a reduction in mass and iron deposition (
Figure 3 and
Figure 4). The patient continued intensive iron chelation therapy with a combination of deferiprone and deferoxamine, resulting in a decreased iron load. Transfusions were maintained every 15 days with the goal of achieving hemoglobin levels above 10 g/dL. The patient achieved complete neurological recovery in the sensory and motor aspects, with only a minor improvement noted in neurogenic bladder dysfunction. Despite our patient experiencing severe myocardial iron overload, she maintained satisfactory systolic function in both the left and right ventricles. There were no observed valvular or conduction abnormalities, and the volume overload resulting from hypertransfusion did not adversely affect her overall cardiac function.
3. Discussion
Extramedullary hematopoiesis is frequently observed as a compensatory response to chronic hemolytic anemia in various hematologic disorders such as thalassemia, primary myelofibrosis (PMF), or following bone marrow radiation. It tends to occur more frequently in men, with a male-to-female ratio of 5:1. Among patients with thalassemia intermedia (NTDT), the incidence of extramedullary hematopoiesis can be as high as 20%, while in those with multi-transfused Thalassemia Major (TDT), the incidence is approximately 1%. Notably, 11–15% of individuals experiencing extramedullary hematopoiesis exhibit a paravertebral location of hematopoietic tissue [
1,
2].
The thoracic region, and to a lesser extent the lumbar region, are the most common sites for extramedullary hematopoiesis. The reason for this predilection is not clearly specified, but in the thoracic area, the subarachnoid space and spinal canal are relatively narrow. Even small intraspinal hematopoietic tissue in this region can lead to spinal compression, unlike in other areas of the spinal cord where larger sizes are required to cause symptoms by exerting excessive pressure. Extramedullary hematopoiesis can manifest in various organs, with associated features such as splenomegaly, hepatomegaly, lymphadenopathy, pleural or pericardial effusions, abdominal effusions, and involvement of the gastrointestinal, genitourinary tract, or lungs [
2,
3]. This involvement can result in symptoms like dysuria and respiratory distress. Central nervous system participation may be linked to increased intracranial pressure, sensory impairment, and motor and sensory deficits, including cord compression.
During fetal development, nearly all organs engage in hematopoiesis, a process that typically ceases after birth [
4]. However, in cases of prolonged chronic anemia accompanied by ineffective erythropoiesis, maintaining the connective tissue supporting extramedullary hematopoiesis becomes crucial for red blood cell production. It is proposed that extramedullary foci may represent remnants of embryonic hematopoietic cells stimulated during periods of chronic anemia and hypoxia. Histologically, extramedullary hematopoiesis typically presents with both immature and mature cells in the erythroid and myeloid series. The presence of dilated sinusoids is notable, responsible for generating inactive red cell precursors. These tissues are susceptible to fatty deposits and fibrosis in a stable state, or they may accumulate extensive iron deposits [
5]. In this specific case, despite being on a transfusion program from an early age, the patient developed extramedullary hematopoiesis with a significant accumulation of iron overload, as observed in the magnetic resonance imaging.
Although history and physical examinations can provide valuable insights, the primary diagnostic assessment of utmost significance is radiographic imaging. This not only broadens the scope of the differential diagnosis but also serves to confirm the presence of hematopoietic tissue. Lesions may be evident on magnetic resonance imaging, particularly showcasing substantial iron deposition in individuals undergoing blood transfusions. Active hematopoietic lesions typically exhibit robust neovascularization, while older, inactive lesions are characterized by the presence of adipose tissue and iron deposition [
6,
7,
8,
9].
Interventions for managing this condition include multiple blood transfusions to suppress erythropoietin production, radiation therapy to inhibit the overgrowth of marrow tissue, surgical decompression, or a combination of these approaches. The relative advantages of these treatments have not been firmly established due to the infrequency of the condition. In thalassemic patients, adequate blood transfusions play a crucial role in reducing inefficient hematopoiesis, thereby mitigating the extensive development of extramedullary hematopoiesis [
10,
11]. It is important to note that complications in hypertransfusion therapy are not uncommon, and efforts should be made to anticipate and prevent them where possible. The target hemoglobin level in hypertransfusion therapy is typically set above 10 g/dL. Additionally, hypertransfusion therapy can serve a dual purpose, being both diagnostic and therapeutic, with a specific response observed in addressing edema and cord compression secondary to extramedullary hematopoiesis.
The literature reports favorable outcomes and effective follow-up assessments of transfusions in treating spinal cord compression resulting from extramedullary hematopoiesis. The primary response is attributed to a reduction in blood flow to these tissues, often preceding any observable decrease in mass volume. Surgical decompression is recommended for managing extramedullary masses, allowing for immediate relief. However, surgical interventions come with several drawbacks, such as the risk of bleeding due to mass vascularization. Other limitations include the potential for incomplete resection and a notable recurrence rate after surgical removal. Furthermore, immediate and complete resection of hematopoietic masses, critical for maintaining adequate hemoglobin levels, may lead to clinical compensation and subsequent deterioration.
Studies have indicated that extramedullary hematopoietic tissue is highly responsive to radiation, with some trials demonstrating significant reduction in mass size and rapid neurological improvement. Reported dosages in the literature range from 900 to 3500 cGy, and after radiation therapy, complete recovery is typically observed [
12]. The main drawbacks of radiotherapy include the absence of a documented histologic diagnosis and the reduction in bone marrow activity associated with radiation exposure. Hydroxyurea (HU), initially approved as an HbF modulator for clinical use in sickle cell anemia patients, has demonstrated significant clinical and laboratory hematological effects in individuals with beta-thalassemia, particularly in those with thalassemia intermedia [
13]. It is known for its minimal side effects. HU is commonly utilized in combination with transfusion or radiotherapy for the treatment of extramedullary hematopoiesis. For patients with an acute presentation unresponsive to adequate radiotherapy or transfusion, laminectomy is currently recommended as an intervention [
14].
Our patient commenced a regimen of blood hypertransfusion, receiving a total of ten units of packed RBCs at a rate of two units per week. Improvement in her condition became apparent after the fourth unit, and her neurological symptoms completely resolved after the tenth unit. Subsequent neurological examinations revealed normal findings, except for a persistent slight brisk reflex in the right knee and Achilles tendon. At that time, her hemoglobin level measured 15 g/dL. Hydroxyurea therapy was introduced to enhance the effectiveness of transfusion in suppressing erythropoiesis without further exacerbating extramedullary hematopoiesis. The combination of transfusion and HU proved to be a successful therapeutic approach, particularly when the potential adverse effects of blood hypertransfusions, such as alloimmunization of red cell antigens and iron overload, needed consideration. The HU treatment initiated at 5 mg/kg per day was gradually increased to 10 mg/kg per day. Unfortunately, the patient experienced bone marrow aplasia with leukopenia and thrombocytopenia, leading to the discontinuation of the treatment.
In the era of emerging treatments for thalassemia patients, it remains to be observed whether these developments will impact the incidence of this potentially hazardous complication within this patient cohort.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}