Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography


Division of Endocrinology and Metabolism, Department of Internal Medicine, Korea University Guro Hospital, Korea University College of Medicine, 148 Gurodong-ro, Guro-gu, Seoul 08308, Korea
*
Author to whom correspondence should be addressed.
Nutrients 2018, 10(10), 1382; https://doi.org/10.3390/nu10101382
Submission received: 13 August 2018
/
Revised: 17 September 2018
/
Accepted: 23 September 2018
/
Published: 28 September 2018
(This article belongs to the Special Issue Diet, Lipid and Lipoprotein Metabolism and Human Health)
{kind=link}
Abstract
:Vascular inflammation plays a central role in atherosclerosis, from initiation and progression to acute thrombotic complications. Modified low-density lipoproteins (LDLs) and apoB-containing particles stimulate plaque inflammation by interacting with macrophages. Loss of function of high-density lipoprotein (HDL) for preventing LDL particles from oxidative modification in dyslipidemic states may amplify modified LDL actions, accelerating plaque inflammation. Diets are one of the most important factors that can affect these processes of lipoprotein oxidation and vascular inflammation. Recently, 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) has emerged as a reliable noninvasive imaging modality for identifying and quantifying vascular inflammation within atherosclerotic lesions based on the high glycolytic activity of macrophages infiltrating active atherosclerotic plaques. Vascular inflammation evaluated by FDG PET has been positively related to metabolic syndrome components and traditional risk factors of cardiovascular disease, including high-sensitivity C-reactive protein, body mass index, and insulin resistance. A positive association of vascular inflammation with endothelial dysfunction, resistin levels, pericardial adipose tissue, and visceral fat area has also been reported. In contrast, HDL cholesterol and adiponectin have been inversely related to vascular inflammation detected by FDG PET. Because of its reproducibility, serial FDG PET shows potential for tracking the effects of dietary interventions and other systemic and local antiatherosclerotic therapies for plaque inflammation.
1. Introduction
Atherosclerotic cardiovascular disease (ASCVD), manifested as various forms of fatal diseases including myocardial infarction, ischemic stroke, and peripheral artery occlusive disease, is the leading cause of morbidity and mortality [1]. Therefore, developing methods for identifying and monitoring the atherosclerotic process in early phases may greatly impact public health. Accumulating data from extensive research indicate that modified lipoproteins and subsequent vascular inflammation are key factors in the atherosclerotic process [2,3]. The oxidation of lipoproteins and vascular inflammation may be significantly influenced by dietary patterns [4,5], suggesting that this may be a link between diet and ASCVD [5,6]. Recently, 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) has attracted attention as an imaging modality for detecting vascular inflammation in atherosclerotic plaques early in relation to these key factors of arteriosclerosis. The main aim of this review is to describe the interactions between lipoprotein and macrophages leading to vascular inflammation during atherosclerosis, effects of diets during this process of inflammation, and evaluation of this vascular inflammation by 18F- FDG PET.
2. Atherosclerosis, an Inflammatory Disease
Inflammation is known to play a key role in the development and progression of atherosclerosis [3], and atherosclerosis is currently considered as a low-grade vascular inflammation [3]. Inflammation contributes to all stages of atherosclerotic cardiovascular disease, from the earliest initiation and evolution of atherosclerosis to acute thrombotic complications [3]. Inflammation not only induces thrombus formation but also inhibits endogenous fibrinolysis such that once formed, the thrombus is firmly maintained [3]. Acute cardiovascular events are thought to be affected mainly by the inflammatory status and constitution of atherosclerotic plaques rather than the extent of vascular stenosis [7].
Activated macrophages are among the major contributors to increased susceptibility of plaque rupture and promoting thrombus formation [3]. Macrophages ingest lipids through the expression of scavenger receptors for modified lipoproteins and develop into foam cells [3] (Figure 1). Physical disruptions of atherosclerotic plaques cause most coronary arterial thrombi that lead to fatal acute myocardial infarction [3]. The activated macrophages in atheroma can secrete proteolytic enzymes, which cause thinning and weakening of the protective fibrous cap of the plaque by degrading collagen in the cap [3]. This makes the plaque more vulnerable to rupture [3]. Activated macrophages can also express tissue factors, which act as the main procoagulants and induce thrombosis in plaques [3].
Elevated circulating inflammatory markers, particularly C-reactive protein (CRP), can predict the risk of atherosclerotic cardiovascular events [3,8] and poor outcomes in acute coronary syndromes [3]. Increasing evidences from population-based studies support that low-grade chronic inflammation, represented by increased levels of CRP, defines the future risk of atherosclerotic complications [3,8]. Therefore, CRP testing may have an important adjunctive role in estimating future cardiovascular risk in addition to the use of traditional risk factors [3,8], and combining CRP and lipid testing to improve cardiovascular risk prediction may have clinical utility [3,8].
Data from experimental and clinical studies indicate that statins not only lower low-density lipoproteins (LDLs) but also reduce plaque inflammation and affect plaque stability [3,9]. Macrophage contents in experimental atherosclerotic plaques can be reduced by pravastatin [3,10,11]. Simvastatin, fluvastatin, and atorvastatin are known to inhibit the expression of tissue factor and matrix metalloproteinases in vivo as well as in vitro [12,13] and appear to attenuate intimal inflammation [14]. Statins may also suppress the expression of adhesion molecules, thus reducing the attachment and adhesion of monocytes to the vascular endothelium [15]. The Cholesterol and Recurrent Events (CARE) investigators reported that CRP levels were lowered by pravastatin independently of the effect of pravastatin on high-density lipoprotein (HDL) or LDL cholesterol [16]. Reduced CRP levels by statins were also demonstrated in clinical studies of lovastatin, simvastatin, and atorvastatin [17,18,19].
CRP testing may be useful for targeted statin therapy, particularly for the primary prevention of cardiovascular events [3]. In the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study, incident cardiovascular disease (CVD) was reduced by 44% by rosuvastatin therapy in apparently healthy persons without hyperlipidemia but with increased high-sensitivity CRP (hsCRP) levels [20]. According to the CARE investigators, the proportion of recurrent coronary events prevented by pravastatin was greater among those with evidence of inflammation, which was defined by the CRP and serum amyloid A levels, compared to those without evidence of inflammation [16,21]. The Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) CRP substudy suggested that CRP screening can be conducted to improve the targeting of statin therapy adjunctively with lipid testing [3,19].
3. Effects of Lipoproteins on Vascular Inflammation
Modified LDL and other apoB-containing particles represent key stimulators of plaque inflammation through interplay with macrophages [2] (Figure 1). The endothelial layers are penetrated by atherogenic lipoproteins at sites of activation, such as branch points of arteries [2,22], and all forms of apoB-containing particles, including LDL, very low-density lipoprotein (VLDL), VLDL remnants, and lipoprotein(a), have the potential for retention within the arterial intima [2,22]. Once retained, LDL may undergo modification by aggregation, lipolysis, oxidation, or proteolysis [2]. According to the oxidation hypothesis of atherosclerosis, this oxidative modification of lipoproteins, primarily LDL, in the vascular wall constitutes a critical event in atherogenesis [2,23]. Modified LDL, frequently including oxidized lipids, chronically stimulates innate and adaptive immune reactions [2,3], resulting in activation of endothelial cells and smooth muscle cells [2,24]. Accordingly, these activated cells express adhesion molecules, chemoattractants, and growth factors to attract circulating monocytes [2,24]. Subsequently, circulating monocytes migrate into the intima and differentiate into macrophages or dendritic cells [2,24]. Intimal monocyte-derived macrophages engulf modified LDLs through their scavenger receptor or other pathways and convert into foam cells packed with droplets of lipoprotein-derived cholesteryl esters [2,25]. These foam cells, exhibiting a proinflammatory phenotype, represent an essential element in the early fatty streak change and play a central role in lesion progression [2]. In these series of processes, it is noteworthy that it is the modified LDL that constitutes the primary driver for foam cell formation [2]. The binding of modified LDL to pattern recognition receptors on macrophages is now thought to be the primary trigger in plaque inflammation [2,22,24,25,26,27].
Loss of the function of HDL to protect LDL particles from oxidative modification in dyslipidemic states, including hypertriglyceridemia, may amplify the role of modified LDL and accelerate intraplaque inflammation [2] (Figure 1). HDL particles exert protective activities against atherosclerosis by effluxing cellular cholesterol, attenuating vascular inflammation, decreasing intracellular oxidative stress and platelet activation, relieving vascular constriction, and sustaining glucose homeostasis [2]. Decreased plasma HDL levels and/or HDL particle numbers may impair the clearance of cholesterol from arterial wall cells, primarily from macrophages and macrophage-derived foam cells [2,3]. HDL particles inhibit chronic inflammation in the arterial wall, which is central in atherosclerosis, through multiple anti-inflammatory actions [2]. HDLs decrease the expression of adhesion molecules in endothelial cells and subsequently suppress monocyte adhesion to the endothelium [2,28,29,30,31,32]. They also reduce monocyte activation through nuclear factor kappa B (NF-κB) and peroxisome proliferator-activated receptor gamma (PPAR-gamma)-dependent pathways [2,33,34,35]. Additionally, apoptosis of both macrophages and endothelial cells by free cholesterol or oxidized LDL loading may be protected by HDL particles [2,36]. Moreover, HDL particles decrease reactive oxygen species generation and intracellular oxidative stress [37,38,39,40,41]. Antioxidant enzymes, including platelet-activating factor acetyl hydrolase and paraoxonase, can also be transported by HDL particles to degrade oxidized lipids and alleviate their proinflammatory effects [3]. Loss of the antioxidative action of HDL in dyslipidemic states can aggravate the build-up of LDL-derived oxidized phospholipids with proinflammatory effects and amplify inflammation in the arterial intima [2,42].
4. Impact of Diets on Inflammation and Lipoprotein Oxidation Modulating Atherosclerotic Process
Effect of diets on LDL oxidation and inflammation has been consistently reported [4,5] and suggested as a possible link between diet and CVD [5,6]. When Mata et al. [4] examined plasma LDL and HDL cholesterol levels, in vitro LDL oxidation, and monocyte adhesion to cultured human endothelial cells in 42 healthy individuals by subjecting them to four consecutive diets with different fat content of saturated fatty acids, monounsaturated fatty acids, and polyunsaturated fatty acids (n-6) and (n-3), a monounsaturated fatty acid-rich diet was associated with more favorable plasma lipid concentrations compared with a saturated fatty acid-rich diet. In addition, this diet was demonstrated to induce a higher resistance of LDL to copper-induced oxidation and a lower monocyte adhesion rate to endothelial cells than other diets with various fatty acid contents [4]. Increased substrate concentration in postprandial states, including both postprandial hypertriglyceridemia and hyperglycemia, are known to generate endothelial dysfunction, one of the earliest indicators of atherosclerosis [5]. The increased level of triglyceride following a high-fat (saturated) diet was inversely related to endothelial function, particularly when accompanied by postprandial hyperglycemia when Nappo et al. [43] explored the effect of various meals with identical calories on endothelial function in participants with and without type 2 diabetes. This impact of postprandial hypertriglyceridemia and hyperglycemia on endothelial function may be mediated by increased oxidative stress accompanied by meal intake, which in turn leads to elevated circulating biomarkers of inflammation and adhesion [5,44]. Inactivation of nitric oxide by increased superoxide production and peroxynitrite generations through combination of superoxide with nitric oxide are considered to be involved in these series of processes [5]. Lipid peroxidation is initiated by the peroxynitrite, which constitutes a potent and long-lasting oxidant with cytotoxic properties [5].
Increasing evidences indicate that induction or alleviation of a proinflammatory condition and subsequent alteration in endothelial function by dietary patterns may be one mechanism that CVD risks are affected by diets [5]. Ingestion of certain macronutrients may induce oxidative stress and inflammatory milieu [5]. Glucose intake is associated with increased superoxide production in leukocytes and mononuclear cells and activation of proinflammatory transcription factors including NF-κB in normal subjects [5]. A high-fat meal promotes endothelial activation, manifested by increased adhesion molecule levels [5,43], and elevation in proinflammatory cytokine interleukin-18 that can weaken plaque stability [5,45]. In contrast, increased consumption of fruits, vegetables, and fibers that are abundant in natural antioxidants largely reduce the adverse effect of high-fat-meal-induced oxidative stress [5]. In addition to this dietary strategy, increasing omega-3 fatty acid and decreasing trans and saturated fatty acids consumption, which are known to prevent coronary heart disease [46], have been related to decreased inflammatory status [5]. Higher trans-fatty acid consumption was associated with increased CRP levels and markers of endothelial dysfunction [47], whereas low-cholesterol/low-saturated fat diets [48] and higher omega-3 fatty acid intake [49] were correlated with reduced plasma CRP concentrations. In a randomized trial with 180 patients with metabolic syndrome, a Mediterranean-style diet significantly reduced vascular inflammatory markers, including serum hsCRP and interleukin-18, and improved insulin resistance and endothelial function [50]. At two years of follow-up, a significant decrease in prevalence of metabolic syndrome was observed in the Mediterranean-style diet group compared to the control subjects [50]. Moreover, Zhong et al. [6] demonstrated that more proinflammatory diets, assessed by a novel dietary inflammatory index score, were independently associated with an increased risk of CVD, cardiovascular, and all-cause mortality in the general population.
Furthermore, some animal studies have tried to evaluate the effect of dietary modification on vascular inflammation and plaque contents of atherosclerotic lesions more directly. Verhamme et al. [51] reported that dietary cholesterol withdrawal induced reduction in LDL, oxidized LDL and CRP levels, restoration of endothelial function, and decrease in lipid, oxidized LDL and macrophage contents of atherosclerotic plaques in miniature pigs. Hartung et al. [52] used histologic and immunohistochemical analysis of the atherosclerotic lesions of rabbit models to show that dietary modification resolved macrophage infiltration, reduced apoptosis of macrophages, and increased smooth muscle cell content within the atherosclerotic plaques.
In clinical testing, there have been trials determining the long-term effect of dietary intervention on markers of vascular inflammation and plaque stability. A 12-month randomized trial with 164 individuals at high risk for CVD showed that enhanced Mediterranean diets attenuated the biomarkers related to atherosclerotic vascular inflammation and plaque vulnerability, including adhesion molecule expression on monocyte surface, CRP, interleukin-6, and endothelial adhesion molecules, compared to a low-fat diet [53]. According to the same group, Mediterranean diets not only improved lipid profiles, including total, LDL, and HDL cholesterol concentrations, but also reduced cellular and plasma inflammatory markers of atherogenesis, such as hsCRP, interleukin-6, and molecules associated with the attraction and adhesion of monocytes to vascular endothelium, compared to a low-fat diet at a longer-term follow-up of three and five years in a randomized controlled trial with high-risk adults without overt CVD [54].
5. Utility of 18F-fluorodeoxyglucose (FDG) Positron Emission Tomography (PET) for Identifying Vascular Inflammation
Recently, 18F-FDG PET has been established as one of the most useful imaging modalities for identifying and measuring inflamed vulnerable atherosclerotic plaques [55]. 18F-FDG PET is generally used in clinical fields to detect 18F-FDG uptake in cells with increased glucose metabolism [56], including inflammatory cells and tumor cells [57]. It has been reported that 18F-FDG accumulation is also increased by active atherosclerosis [56,58,59]. Inflammatory cell infiltration, particularly of macrophages, and subendothelial proliferation of smooth muscle cells and macrophages within atherosclerotic foci are considered to be highly associated with 18F-FDG uptake on PET images [60,61]. Visualization of vascular inflammation by 18F-FDG PET is attributed to the high glycolytic activity of macrophages within active atherosclerotic plaques [56,62] (Figure 1). Rudd et al. [63] reported that inflammatory plaques were identified by 18F-FDG PET in eight patients followed by carotid endarterectomy. Rogers et al. [64] described that 18F-FDG PET showed a higher uptake within the ascending aorta, left main coronary artery, and the culprit lesion in 10 patients with recent acute coronary syndrome compared to 15 individuals with stable angina. In the preclinical testing, Ogawa et al. [65] demonstrated that 18F-FDG accumulation in atherosclerotic lesions was ascribable to macrophages in a rabbit model of atherosclerosis. According to Tawakol et al. [66], 18F-FDG PET signals from carotid plaques were significantly correlated with macrophage staining of corresponding pathologic specimens isolated after endarterectomy. These studies indicate that 18F-FDG PET is a useful noninvasive tool for detecting and quantifying inflammation within atherosclerotic lesions, particularly macrophage-associated vascular inflammation [60,67].
Moreover, because of the high reproducibility of 18F-FDG uptake measurements [60,68,69], PET scanning may be useful for tracking the effects of atherosclerosis treatment [69]. Tahara et al. [60] reported good reproducibility and intraobserver and interobserver variability of less than 5% in standardized uptake value (SUV) measurements of plaque inflammation. The SUV is calculated as the decay-corrected tissue concentration of FDG divided by the administered dose per body weight [69]. The mean and maximum blood-normalized SUVs, known as the target-to-background ratio (TBR), are widely used to measure vascular 18F-FDG uptake [69]. Both the mean and maximum TBR have been reported to be equally reproducible [69]. Rudd et al. [69] suggested that systemic arterial therapies can be tracked by the mean TBR, whereas the maximum TBR can be used to monitor local, plaque-based therapy. In summary, serial 18F-FDG PET imaging may be helpful for monitoring the plaque burden and inflammatory activity [60] and used to examine responses in vascular inflammation to medical or local therapy because of its reproducibility [69].
6. Association of Vascular Inflammation Assessed by Positron Emission Tomography (PET) with Markers of Lipoprotein Metabolism and other Risk Factors for Atherosclerotic Cardiovascular Disease (ASCVD)
The association of vascular inflammation with markers of lipoprotein metabolism and other risk factors for ASCVD has been explored using 18F-FDG PET imaging in previous studies. A positive relationship between components of metabolic syndrome or traditional risk factors for ASCVD and vascular inflammation has been demonstrated [70]. Vascular inflammation quantified by FDG uptake in carotid atherosclerosis was positively related to waist circumference, hypertensive medication, and homeostasis model assessment of insulin resistance (HOMA-IR) and inversely associated with HDL cholesterol in 216 subjects who underwent cancer screening [70]. In the same study, age- and gender-adjusted FDG accumulation in carotid artery was increased in proportion to the satisfied number of metabolic syndrome components, suggesting an association between metabolic syndrome and vascular inflammation [70]. Additionally, hypercholesterolemia showed consistent positive correlations with FDG uptake in three different arteries, including the abdominal aorta, lilac, and proximal femoral arteries of 156 patients [61]. Studies confirmed a significant positive correlation of vascular inflammation with total cholesterol [56], blood pressure [71], body mass index (BMI) [71], waist circumference [70,71], waist-to-hip ratio [71], and insulin resistance [70]. A negative relationship between vascular 18F-FDG index and HDL cholesterol was consistently observed in other studies [56,70].
Inflammation within atherosclerotic plaques detected by 18F-FDG PET has been positively correlated with the circulating inflammatory marker hsCRP [70,71,72]. Vascular inflammation investigated by PET/computed tomography (CT) was reported to be increased in healthy individuals without hyperlipidemia but with high hsCRP values, suggesting that 18F-FDG uptake reflects early inflammatory changes in vascular walls in low-risk individuals [72]. In the same study, hsCRP and diastolic blood pressure were found to be independent determinants of vascular inflammation represented by maximal TBR [72]. Other studies also confirmed a significant positive relationship between hsCRP and vascular inflammation measured by FDG uptake [70,71,73].
Honda et al. confirmed that vascular inflammation in carotid arteries assessed by 18F-FDG PET was independently associated with endothelial dysfunction represented by a decreased percentage (%) flow-mediated dilation of the brachial arteries in 145 adults [74]. Endothelial dysfunction is one of the earliest steps in the atherosclerotic process and was reported to be correlated with several cardiovascular risk factors, including dyslipidemia, smoking status, obesity, insulin resistance, diabetes, hypertension, and CRP [74].
The association between vascular inflammation and serum adiponectin or resistin levels has also been investigated. Adiponectin is a metabolically active adipokine [73] that exerts protective effects against the development of insulin resistance, inflammation, and atherosclerosis [73,75]. In contrast, resistin was shown to induce adhesion molecules [76] and foam cell formation in experimental studies [77], and has been reported to play a role in obesity-associated subclinical inflammation, atherosclerosis, and CVD [78]. In contrast to rodents in which resistin is nearly exclusively derived from adipose tissue [79], in humans, resistin is abundantly expressed in inflammatory cells, particularly macrophages, which are important in inflammation and atherosclerosis [80]. In our previous study [73], serum adiponectin levels were negatively correlated with vascular inflammation represented by the mean TBR, while resistin levels were positively correlated with the same variable. In this study, vascular inflammation was independently associated with resistin levels even after adjusting for other cardiovascular risk factors, such as hsCRP [73].
Additionally, pericardial adipose tissue and visceral fat area were shown to be associated with vascular inflammation measured by 18F-FDG PET in 93 men and women without diabetes or CVD [81]. Pericardial adipose tissue and visceral fat area were also positively associated with major cardiovascular risk factors, including systolic blood pressure, LDL cholesterol, triglycerides, glucose, insulin resistance represented by HOMA-IR, and hsCRP, while they were inversely associated with HDL cholesterol [81].
7. Improvement in Vascular Inflammation Assessed by Positron Emission Tomography (PET) by the Medical Manage and Lifestyle Intervention Including Diet Control
Vascular inflammation may denote a dynamic aspect of atherosclerotic lesions, which varies during the course of atherosclerosis [82]. 18F-FDG uptake by atherosclerotic plaques is considered a transient phenomenon that is attenuated according to decreases in active inflammatory components [56]. Clinical and animal studies have explored whether vascular inflammation visualized by PET/CT can be reversed by interventions, including lifestyle modification and pharmacological therapy [56,60,83,84]. The antioxidant probucol was shown to decrease macrophage-rich fatty-streak lesions of atherosclerosis in Watanabe rabbits [85]. Ogawa et al. [84] reported that probucol treatment significantly reduced macrophage infiltration and 18F-FDG uptake by aortas, whereas intimal thickening was not altered in Watanabe heritable hyperlipidemic rabbits.
PPAR-gamma receptors are expressed by cells involved in the atherosclerotic process, including endothelial cells, smooth muscle cells, T-lymphocytes, and primarily macrophages [86,87]. In animal studies, thiazolidinediones, which are synthetic PPAR-gamma receptor ligands, were shown to have potent antiatherosclerotic properties suppressing the formation of atherosclerotic plaques, in addition to their antidiabetic effects [88,89]. Pioglitazone, a thiazolidinedione, was demonstrated to have vascular benefits [90,91] and preventive effects on all-cause mortality, myocardial infarction, and stroke in diabetic patients [92]. In this background, Vucic et al. [83] demonstrated that pioglitazone arrested the progression of vascular inflammation evaluated by 18F-FDG PET/CT in atherosclerotic rabbits. In the same study, immunoreactivity for macrophages and oxidized phospholipids from the aortas showed a significantly decreased value in the pioglitazone group compared to that in control rabbits [83].
Tahara et al. [60] reported that simvastatin therapy for three months reduced plaque inflammation as represented by decreased 18F-FDG uptakes in FDG-PET in humans. In the simvastatin group, the reduction in 18F-FDG accumulation was well correlated with the increase in HDL cholesterol but not with the decrease in LDL cholesterol [60], suggesting that an LDL-cholesterol-independent mechanism may be involved in the anti-inflammatory action of simvastatin on atherosclerotic plaques [60].
When analyzed with in vivo PET/CT scan and ex vivo gamma counting of excised aorta in hypercholesterolemic mice deficient in LDL receptor and expressing only apolipoprotein B-100, 18F-FDG uptake in the aorta was significantly lowered by a cholesterol-lowering diet [93]. At the same time, this dietary intervention effectively reduced plaque burden and macrophage count within atherosclerotic lesions on aortic histopathology [93]. Notably in a clinical setting, Lee et al. [56] reported that atherogenic risk reduction through lifestyle intervention reversed vascular 18F-FDG uptake in PET/CT in 60 healthy adults, and the extent of reduction in the 18F-FDG index correlated well with elevations in plasma HDL cholesterol levels [56]. Lifestyle modification strategy in this study [56] included individual dietary counseling provided by a registered dietitian. These results indicate that FDG-PET can also be used to track the effect of diet management, which is an important factor affecting lipoprotein oxidation and endothelial function, on plaque inflammation.
The results of previous studies suggest the utility of 18F-FDG PET for monitoring the effects of antiatherosclerotic therapies, including dietary intervention on vascular inflammation. Serial 18F-FDG PET may be a noninvasive tool for tracking the effects of therapeutic interventions on plaque inflammation and developing novel drugs that can recede inflammation on vulnerable plaques [56,60,83,84].
8. Conclusions
An increasing body of evidence supports that vascular inflammation, which is stimulated by modified LDLs and apoB-containing particles, and their interplay with macrophages plays a key action in atherosclerosis. 18F-FDG PET has emerged as a reliable imaging technique capable of identifying vascular inflammation associated with macrophage infiltration. Clinical studies suggest the utility of 18F-FDG PET for monitoring effects of dietary interventions and other antiatherosclerotic therapies on plaque inflammation. Although experimental and clinical studies provide indirect evidences that vascular inflammation may be affected by diets, further clinical studies addressing the effect of diet directly on atherosclerotic vascular inflammation and plaque vulnerability are needed. 18F-FDG PET can be a useful tool in identifying this issue.
Author Contributions
Conceptualization, K.M.C. and Y.-B.L.; Writing—Original Draft Preparation, Y.-B.L.; Writing—Review & Editing, K.M.C.; Supervision, K.M.C.
Funding
This study was supported by a grant from Korea University (K1809301, Q1625561).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics--2015 update: A report from the american heart association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Rached, F.; Kontush, A.; Chapman, M.J. Impact of lipoproteins on atherobiology: Emerging insights. Cardiol. Clin. 2018, 36, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Mata, P.; Alonso, R.; Lopez-Farre, A.; Ordovas, J.M.; Lahoz, C.; Garces, C.; Caramelo, C.; Codoceo, R.; Blazquez, E.; de Oya, M. Effect of dietary fat saturation on ldl oxidation and monocyte adhesion to human endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Giugliano, D. Diet and inflammation: A link to metabolic and cardiovascular diseases. Eur. Heart J. 2006, 27, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Guo, L.; Zhang, L.; Li, Y.; He, R.; Cheng, G. Inflammatory potential of diet and risk of cardiovascular disease or mortality: A meta-analysis. Sci. Rep. 2017, 7, 6367. [Google Scholar] [CrossRef] [PubMed]
- Van Lennep, J.E.; Westerveld, H.T.; van Lennep, H.W.; Zwinderman, A.H.; Erkelens, D.W.; van der Wall, E.E. Apolipoprotein concentrations during treatment and recurrent coronary artery disease events. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2408–2413. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. High-sensitivity C-reactive protein: Potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001, 103, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Bui, A.V.; Diesch, J.; Manasseh, R.; Hausding, C.; Rivera, J.; Haviv, I.; Agrotis, A.; Htun, N.M.; Jowett, J.; et al. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microrna expression profiling. Circ. Res. 2013, 113, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Sukhova, G.K.; Herrington, D.M.; Libby, P. Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. J. Am. Coll. Cardiol. 1998, 31, 684–691. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Libby, P.; Rabkin, E.; Hill, C.C.; Enomoto, M.; Hirouchi, Y.; Shiomi, M.; Aikawa, M. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation 2001, 103, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.; Zhou, H.; Ye, H.; Li, J. Atorvastatin lowers plasma matrix metalloproteinase-9 in patients with acute coronary syndrome. Clin. Chem. 2004, 50, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Bellosta, S.; Via, D.; Canavesi, M.; Pfister, P.; Fumagalli, R.; Paoletti, R.; Bernini, F. Hmg-coa reductase inhibitors reduce mmp-9 secretion by macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Bustos, C.; Hernandez-Presa, M.A.; Ortego, M.; Tunon, J.; Ortega, L.; Perez, F.; Diaz, C.; Hernandez, G.; Egido, J. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J. Am. Coll. Cardiol. 1998, 32, 2057–2064. [Google Scholar] [CrossRef] [Green Version]
- Niwa, S.; Totsuka, T.; Hayashi, S. Inhibitory effect of fluvastatin, an HMG-CoA reductase inhibitor, on the expression of adhesion molecules on human monocyte cell line. Int. J. Immunopharmacol. 1996, 18, 669–675. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.A.; Sacks, F.; Braunwald, E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The cholesterol and recurrent events (care) investigators. Circulation 1999, 100, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.A.; Danielson, E.; Rifai, N.; Ridker, P.M. Effect of statin therapy on c-reactive protein levels: The pravastatin inflammation/CRP evaluation (PRINCE): A randomized trial and cohort study. JAMA 2001, 286, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Stein, D.; Balis, D.; Grundy, S.M.; Adams-Huet, B.; Devaraj, S. Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation 2001, 103, 1933–1935. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Clearfield, M.; Downs, J.R.; Weis, S.E.; Miles, J.S.; Gotto, A.M., Jr. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N. Eng. J. Med. 2001, 344, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N. Eng. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.A.; Sacks, F.M.; Moye, L.A.; Goldman, S.; Flaker, G.C.; Braunwald, E. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events (CARE) investigators. Circulation 1998, 98, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Eng. J. Med. 1989, 320, 915–924. [Google Scholar]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Witztum, J.L. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2311–2316. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; Sposito, A.C. Hypertension and dyslipidaemia in obesity and insulin resistance: Pathophysiology, impact on atherosclerotic disease and pharmacotherapy. Pharmacol. Ther. 2008, 117, 354–373. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Spillmann, F.; Lorenz, M.; Meloni, M.; Jacobs, F.; Egorova, M.; Stangl, V.; De Geest, B.; Schultheiss, H.P.; Tschope, C. Vascular-protective effects of high-density lipoprotein include the downregulation of the angiotensin II type 1 receptor. Hypertension 2009, 53, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.A.; Bobik, A.; Murphy, A.; Kanellakis, P.; Blombery, P.; Mukhamedova, N.; Woollard, K.; Lyon, S.; Sviridov, D.; Dart, A.M. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ. Res. 2008, 103, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Van Leuven, S.I.; Birjmohun, R.S.; Franssen, R.; Bisoendial, R.J.; de Kort, H.; Levels, J.H.; Basser, R.L.; Meijers, J.C.; Kuivenhoven, J.A.; Kastelein, J.J.; et al. ApoAI-phosphatidylcholine infusion neutralizes the atherothrombotic effects of C-reactive protein in humans. J. Thromb. Haemost. 2009, 7, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockerill, G.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, L.; Franceschini, G.; Sirtori, C.R.; De Palma, A.; Saresella, M.; Ferrante, P.; Taramelli, D. Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem. Biophys. Res. Commun. 1997, 238, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J. Lipid Res. 1999, 40, 345–353. [Google Scholar] [PubMed]
- Bursill, C.A.; Castro, M.L.; Beattie, D.T.; Nakhla, S.; van der Vorst, E.; Heather, A.K.; Barter, P.J.; Rye, K.A. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Dousset, N.; Ferretti, G.; Bacchetti, T.; Curatola, G.; Salvayre, R. Antioxidant and cytoprotective properties of high-density lipoproteins in vascular cells. Free Radic. Biol. Med. 2006, 41, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Speidel, M.T.; Booyse, F.M.; Abrams, A.; Moore, M.A.; Chung, B.H. Lipolyzed hypertriglyceridemic serum and triglyceride-rich lipoprotein cause lipid accumulation in and are cytotoxic to cultured human endothelial cells. High density lipoproteins inhibit this cytotoxicity. Thromb. Res. 1990, 58, 251–264. [Google Scholar] [CrossRef]
- Hamilton, K.K.; Zhao, J.; Sims, P.J. Interaction between apolipoproteins A-Iand A-II and the membrane attack complex of complement. Affinity of the apoproteins for polymeric C9. J. Biol. Chem. 1993, 268, 3632–3638. [Google Scholar] [PubMed]
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Negre-Salvayre, A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized HDL. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Robbesyn, F.; Garcia, V.; Auge, N.; Vieira, O.; Frisach, M.F.; Salvayre, R.; Negre-Salvayre, A. HDL counterbalance the proinflammatory effect of oxidized HDL by inhibiting intracellular reactive oxygen species rise, proteasome activation, and subsequent NF-kappaB activation in smooth muscle cells. FASEB J. 2003, 17, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Ansell, B.J.; Navab, M.; Hama, S.; Kamranpour, N.; Fonarow, G.; Hough, G.; Rahmani, S.; Mottahedeh, R.; Dave, R.; Reddy, S.T.; et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 2003, 108, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- Nappo, F.; Esposito, K.; Cioffi, M.; Giugliano, G.; Molinari, A.M.; Paolisso, G.; Marfella, R.; Giugliano, D. Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: Role of fat and carbohydrate meals. J. Am. Coll. Cardiol. 2002, 39, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Bowen, P.E.; Borthakur, G. Postprandial lipid oxidation and cardiovascular disease risk. Curr. Atheroscler. Rep. 2004, 6, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Nappo, F.; Giugliano, F.; Di Palo, C.; Ciotola, M.; Barbieri, M.; Paolisso, G.; Giugliano, D. Meal modulation of circulating interleukin 18 and adiponectin concentrations in healthy subjects and in patients with type 2 diabetes mellitus. Am. J. Clin. Nutr. 2003, 78, 1135–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.B.; Willett, W.C. Optimal diets for prevention of coronary heart disease. JAMA 2002, 288, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, E.; Schulze, M.B.; Meigs, J.B.; Manson, J.E.; Rifai, N.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Consumption of trans fatty acids is related to plasma biomarkers of inflammation and endothelial dysfunction. J. Nutr. 2005, 135, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Schillaci, G.; Savarese, G.; Gemelli, F.; Mannarino, M.R.; Siepi, D.; Bagaglia, F.; Mannarino, E. Attenuation of inflammation with short-term dietary intervention is associated with a reduction of arterial stiffness in subjects with hypercholesterolaemia. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, E.; Schulze, M.B.; Manson, J.E.; Meigs, J.B.; Albert, C.M.; Rifai, N.; Willett, W.C.; Hu, F.B. Consumption of (n-3) fatty acids is related to plasma biomarkers of inflammation and endothelial activation in women. J. Nutr. 2004, 134, 1806–1811. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Marfella, R.; Ciotola, M.; Di Palo, C.; Giugliano, F.; Giugliano, G.; D’Armiento, M.; D’Andrea, F.; Giugliano, D. Effect of a mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: A randomized trial. JAMA 2004, 292, 1440–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhamme, P.; Quarck, R.; Hao, H.; Knaapen, M.; Dymarkowski, S.; Bernar, H.; Van Cleemput, J.; Janssens, S.; Vermylen, J.; Gabbiani, G.; et al. Dietary cholesterol withdrawal reduces vascular inflammation and induces coronary plaque stabilization in miniature pigs. Cardiovasc. Res. 2002, 56, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Hartung, D.; Sarai, M.; Petrov, A.; Kolodgie, F.; Narula, N.; Verjans, J.; Virmani, R.; Reutelingsperger, C.; Hofstra, L.; Narula, J. Resolution of apoptosis in atherosclerotic plaque by dietary modification and statin therapy. J. Nucl. Med. 2005, 46, 2051–2056. [Google Scholar] [PubMed]
- Casas, R.; Sacanella, E.; Urpi-Sarda, M.; Chiva-Blanch, G.; Ros, E.; Martinez-Gonzalez, M.A.; Covas, M.I.; Salas-Salvado, J.; Fiol, M.; Aros, F.; et al. The effects of the mediterranean diet on biomarkers of vascular wall inflammation and plaque vulnerability in subjects with high risk for cardiovascular disease. A randomized trial. PLoS ONE 2014, 9, e100084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, R.; Sacanella, E.; Urpi-Sarda, M.; Corella, D.; Castaner, O.; Lamuela-Raventos, R.M.; Salas-Salvado, J.; Martinez-Gonzalez, M.A.; Ros, E.; Estruch, R. Long-term immunomodulatory effects of a mediterranean diet in adults at high risk of cardiovascular disease in the prevencion con Dieta mediterranea (PREDIMED) randomized controlled trial. J. Nutr. 2016, 146, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haim, S.; Kupzov, E.; Tamir, A.; Israel, O. Evaluation of 18F-FDG uptake and arterial wall calcifications using 18F-FDG PET/CT. J. Nucl. Med. 2004, 45, 1816–1821. [Google Scholar] [PubMed]
- Lee, S.J.; On, Y.K.; Lee, E.J.; Choi, J.Y.; Kim, B.T.; Lee, K.H. Reversal of vascular 18F-FDG uptake with plasma high-density lipoprotein elevation by atherogenic risk reduction. J. Nucl. Med. 2008, 49, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Delbeke, D. Oncological applications of FDG pet imaging. J. Nucl. Med. 1999, 40, 1706–1715. [Google Scholar] [PubMed]
- Tatsumi, M.; Cohade, C.; Nakamoto, Y.; Wahl, R.L. Fluorodeoxyglucose uptake in the aortic wall at PET/CT: Possible finding for active atherosclerosis. Radiology 2003, 229, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, M.P.; Freiman, A.; Larson, S.M.; Strauss, H.W. Association of vascular 18F-FDG uptake with vascular calcification. J. Nucl. Med. 2005, 46, 1278–1284. [Google Scholar] [PubMed]
- Tahara, N.; Kai, H.; Ishibashi, M.; Nakaura, H.; Kaida, H.; Baba, K.; Hayabuchi, N.; Imaizumi, T. Simvastatin attenuates plaque inflammation: Evaluation by fluorodeoxyglucose positron emission tomography. J. Am. Coll. Cardiol. 2006, 48, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Jang, S.; Cucchiara, A.; Newberg, A.B.; Alavi, A. 18F FDG uptake in the large arteries: A correlation study with the atherogenic risk factors. Semin. Nucl. Med. 2002, 32, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Kubota, K.; Kubota, R.; Ido, T.; Tamahashi, N. High accumulation of fluorine-18-fluorodeoxyglucose in turpentine-induced inflammatory tissue. J. Nucl. Med. 1995, 36, 1301–1306. [Google Scholar] [PubMed]
- Rudd, J.H.; Warburton, E.A.; Fryer, T.D.; Jones, H.A.; Clark, J.C.; Antoun, N.; Johnstrom, P.; Davenport, A.P.; Kirkpatrick, P.J.; Arch, B.N.; et al. Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation 2002, 105, 2708–2711. [Google Scholar] [CrossRef] [PubMed]
- Rogers, I.S.; Nasir, K.; Figueroa, A.L.; Cury, R.C.; Hoffmann, U.; Vermylen, D.A.; Brady, T.J.; Tawakol, A. Feasibility of FDG imaging of the coronary arteries: Comparison between acute coronary syndrome and stable angina. JACC Cardiovasc. Imaging 2010, 3, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Ishino, S.; Mukai, T.; Asano, D.; Teramoto, N.; Watabe, H.; Kudomi, N.; Shiomi, M.; Magata, Y.; Iida, H.; et al. (18)F-FDG accumulation in atherosclerotic plaques: Immunohistochemical and pet imaging study. J. Nucl. Med. 2004, 45, 1245–1250. [Google Scholar] [PubMed]
- Tawakol, A.; Migrino, R.Q.; Bashian, G.G.; Bedri, S.; Vermylen, D.; Cury, R.C.; Yates, D.; LaMuraglia, G.M.; Furie, K.; Houser, S.; et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J. Am. Coll. Cardiol. 2006, 48, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Kai, H.; Nakaura, H.; Mizoguchi, M.; Ishibashi, M.; Kaida, H.; Baba, K.; Hayabuchi, N.; Imaizumi, T. The prevalence of inflammation in carotid atherosclerosis: Analysis with fluorodeoxyglucose-positron emission tomography. Eur. Heart J. 2007, 28, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.H.; Myers, K.S.; Bansilal, S.; Machac, J.; Rafique, A.; Farkouh, M.; Fuster, V.; Fayad, Z.A. (18)fluorodeoxyglucose positron emission tomography imaging of atherosclerotic plaque inflammation is highly reproducible: Implications for atherosclerosis therapy trials. J. Am. Coll. Cardiol. 2007, 50, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Rudd, J.H.; Myers, K.S.; Bansilal, S.; Machac, J.; Pinto, C.A.; Tong, C.; Rafique, A.; Hargeaves, R.; Farkouh, M.; Fuster, V.; et al. Atherosclerosis inflammation imaging with 18F-FDG pet: Carotid, iliac, and femoral uptake reproducibility, quantification methods, and recommendations. J. Nucl. Med. 2008, 49, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Kai, H.; Yamagishi, S.; Mizoguchi, M.; Nakaura, H.; Ishibashi, M.; Kaida, H.; Baba, K.; Hayabuchi, N.; Imaizumi, T. Vascular inflammation evaluated by [18F]-fluorodeoxyglucose positron emission tomography is associated with the metabolic syndrome. J. Am. Coll. Cardiol. 2007, 49, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Yong, H.S.; Hwang, S.Y.; Eo, J.S.; Hong, H.C.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, D.S.; Baik, S.H.; et al. Association of pooled cohort risk scores with vascular inflammation and coronary artery calcification in Korean adults. Metabolism 2016, 65, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Kim, S.; Park, M.S.; Yang, S.J.; Kim, T.N.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Seo, H.S.; Baik, S.H.; et al. Vascular inflammation stratified by C-reactive protein and low-density lipoprotein cholesterol levels: Analysis with 18F-FDG pet. J. Nucl. Med. 2011, 52, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Kim, S.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Association of adiponectin, resistin, and vascular inflammation: Analysis with 18F-fluorodeoxyglucose positron emission tomography. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Tahara, N.; Nitta, Y.; Tahara, A.; Igata, S.; Bekki, M.; Nakamura, T.; Sugiyama, Y.; Kaida, H.; Kurata, S.; et al. Vascular inflammation evaluated by [18F]-fluorodeoxyglucose-positron emission tomography/computed tomography is associated with endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y. Adiponectin: Identification, physiology and clinical relevance in metabolic and vascular disease. Atheroscler. Suppl. 2005, 6, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Maemura, K.; Takeda, N.; Harada, T.; Nojiri, T.; Imai, Y.; Manabe, I.; Utsunomiya, K.; Nagai, R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: A new insight into adipocytokine-endothelial cell interactions. Biochem. Biophys. Res. Commun. 2004, 314, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Lin, C.Y.; Tsai, J.Y.; Wu, Y.L.; Su, K.H.; Lu, K.Y.; Hsiao, S.H.; Pan, C.C.; Kou, Y.R.; Hsu, Y.P.; et al. Resistin increases lipid accumulation by affecting class A scavenger receptor, CD36 and ATP-binding cassette transporter-A1 in macrophages. Life Sci. 2009, 84, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Filkova, M.; Haluzik, M.; Gay, S.; Senolt, L. The role of resistin as a regulator of inflammation: Implications for various human pathologies. Clin. Immunol. 2009, 133, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Buckels, A.C.; Kinghorn, I.J.; Murdock, P.R.; Holbrook, J.D.; Plumpton, C.; Macphee, C.H.; Smith, S.A. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem. Biophys. Res. Commun. 2003, 300, 472–476. [Google Scholar] [CrossRef]
- Hong, H.C.; Hwang, S.Y.; Park, S.; Ryu, J.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Implications of pericardial, visceral and subcutaneous adipose tissue on vascular inflammation measured using 18FDG-PET/CT. PLoS ONE 2015, 10, e0135294. [Google Scholar] [CrossRef] [PubMed]
- Cocker, M.S.; Mc Ardle, B.; Spence, J.D.; Lum, C.; Hammond, R.R.; Ongaro, D.C.; McDonald, M.A.; Dekemp, R.A.; Tardif, J.C.; Beanlands, R.S. Imaging atherosclerosis with hybrid [18F]fluorodeoxyglucose positron emission tomography/computed tomography imaging: What leonardo da vinci could not see. J. Nucl. Med. 2012, 19, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Vucic, E.; Dickson, S.D.; Calcagno, C.; Rudd, J.H.; Moshier, E.; Hayashi, K.; Mounessa, J.S.; Roytman, M.; Moon, M.J.; Lin, J.; et al. Pioglitazone modulates vascular inflammation in atherosclerotic rabbits noninvasive assessment with FDG-PET-CT and dynamic contrast-enhanced mr imaging. JACC Cardiovasc. Imaging 2011, 4, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Magata, Y.; Kato, T.; Hatano, K.; Ishino, S.; Mukai, T.; Shiomi, M.; Ito, K.; Saji, H. Application of 18F-FDG pet for monitoring the therapeutic effect of antiinflammatory drugs on stabilization of vulnerable atherosclerotic plaques. J. Nucl. Med. 2006, 47, 1845–1850. [Google Scholar] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E. In vivo inhibition of foam cell development by probucol in watanabe rabbits. Am. J. Cardiol. 1988, 62, 6B–12B. [Google Scholar] [CrossRef]
- Marx, N.; Mach, F.; Sauty, A.; Leung, J.H.; Sarafi, M.N.; Ransohoff, R.M.; Libby, P.; Plutzky, J.; Luster, A.D. Peroxisome proliferator-activated receptor-gamma activators inhibit IFN-gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells. J. Immunol. 2000, 164, 6503–6508. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Kitami, Y.; Yang, Z.H.; Nakamura, M.; Okura, T.; Hiwada, K. Vascular inflammation is negatively autoregulated by interaction between CCAAT/enhancer-binding protein-delta and peroxisome proliferator-activated receptor-gamma. Circ. Res. 2002, 91, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Meehan, W.P.; Kintscher, U.; Jackson, S.; Wakino, S.; Noh, G.; Palinski, W.; Hsueh, W.A.; Law, R.E. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Brown, K.K.; Silvestre, M.J.; Willson, T.M.; Palinski, W.; Glass, C.K. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J. Clin. Investig. 2000, 106, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, T.; Meyer, P.M.; Feinstein, S.B.; Davidson, M.H.; Kondos, G.T.; D’Agostino, R.B., Sr.; Perez, A.; Provost, J.C.; Haffner, S.M. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: A randomized trial. JAMA 2006, 296, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A.; Jure, H.; De Larochelliere, R.; Staniloae, C.S.; Mavromatis, K.; et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefebvre, P.J.; Murray, G.D.; et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the proactive study (PROspective pioglitazone clinical trial in macrovascular events): A randomised controlled trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Hellberg, S.; Sippola, S.; Liljenback, H.; Virta, J.; Silvola, J.M.U.; Stahle, M.; Savisto, N.; Metso, J.; Jauhiainen, M.; Saukko, P.; et al. Effects of atorvastatin and diet interventions on atherosclerotic plaque inflammation and [18F]FDG uptake in Ldlr-/-Apob100/100mice. Atherosclerosis 2017, 263, 369–376. [Google Scholar] [CrossRef] [PubMed]
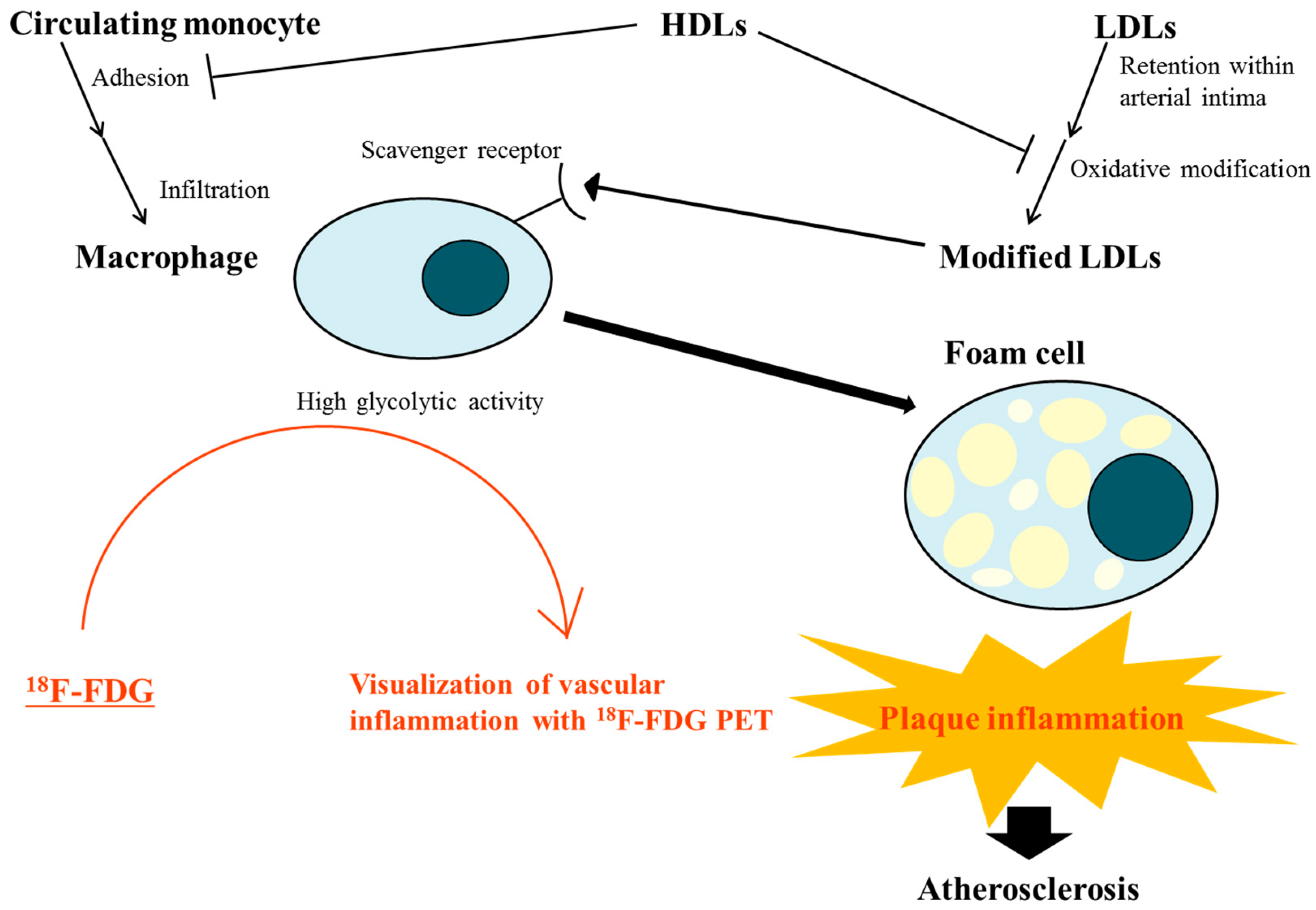
Figure 1.
Interplay of lipoproteins and macrophages linked to plaque inflammation during atherosclerotic process and evaluation of this vascular inflammation by 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET). Modified low-density lipoproteins (LDLs) trigger plaque inflammation by interacting with macrophages. The macrophages ingest modified LDL through scavenger receptors and evolve into foam cells, which play a key role in the development and progression of atherosclerosis. High-density lipoproteins (HDLs) protect LDLs from oxidative modification and inhibit monocyte adhesion to the endothelium by decreasing the expression of adhesion molecules in endothelial cells. Loss of this protective function of HDL in dyslipidemic states may accelerate plaque inflammation, amplifying the role of modified LDLs. The high glycolytic activity of infiltrated macrophages enables visualization of vascular inflammation in atherosclerotic lesions by 18F-FDG PET.
Figure 1.
Interplay of lipoproteins and macrophages linked to plaque inflammation during atherosclerotic process and evaluation of this vascular inflammation by 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET). Modified low-density lipoproteins (LDLs) trigger plaque inflammation by interacting with macrophages. The macrophages ingest modified LDL through scavenger receptors and evolve into foam cells, which play a key role in the development and progression of atherosclerosis. High-density lipoproteins (HDLs) protect LDLs from oxidative modification and inhibit monocyte adhesion to the endothelium by decreasing the expression of adhesion molecules in endothelial cells. Loss of this protective function of HDL in dyslipidemic states may accelerate plaque inflammation, amplifying the role of modified LDLs. The high glycolytic activity of infiltrated macrophages enables visualization of vascular inflammation in atherosclerotic lesions by 18F-FDG PET.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, Y.-B.; Choi, K.M. Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography. Nutrients 2018, 10, 1382. https://doi.org/10.3390/nu10101382
AMA Style
Lee Y-B, Choi KM. Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography. Nutrients. 2018; 10(10):1382. https://doi.org/10.3390/nu10101382
Chicago/Turabian StyleLee, You-Bin, and Kyung Mook Choi. 2018. "Diet-Modulated Lipoprotein Metabolism and Vascular Inflammation Evaluated by 18F-fluorodeoxyglucose Positron Emission Tomography" Nutrients 10, no. 10: 1382. https://doi.org/10.3390/nu10101382
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.