1. Introduction
The use of agents that induce interstrand crosslinks (ICLs), notably the platinum-based analogues, remains a mainstay of cancer treatment and they are used to treat a variety of cancer types including lung, ovarian, and head and neck cancers. Platinum-based drugs induce DNA damage by forming a variety of DNA lesions including ICLs, intrastrand crosslinks, and monoadducts. While platinum-based chemotherapy is often initially effective, resistance to these drugs usually occurs. Resistance to platinums has been attributed to several mechanisms including decreased drug accumulation, loss of mismatch and base excision repair, increased translesion synthesis as well as increased DNA repair [
1]. Extensive work devoted to understanding mechanisms of resistance to these therapies has led to the identification of novel drug targets for enhancing therapeutic response and overcoming chemoresistance, including targeting the 5′-3′ structure-specific endonuclease ERCC1/XPF (Excision Repair Cross-Complementation Group 1/Xeroderma Pigmentosum Group F) [
2,
3]. ERCC1/XPF is a critical complex involved in the repair of DNA ICLs with essential functions in both replication-independent and -dependent ICL repair pathways [
4,
5]. As such, it is well-established that ERCC1 expression is altered in several tumor types, including lung, head and neck, and ovarian cancers, suggesting its potential use as a drug target or biomarker for response to platinum-based chemotherapy. Indeed, this possibility has been thoroughly investigated in preclinical and clinical studies. Clinical data have shown that low ERCC1 expression is associated with increased overall survival in response to chemotherapy [
6]. Further data showed that low ERCC1 expression was associated with a positive response to platinum-based chemotherapy in non-small cell lung cancer patients [
7]. Some mixed results have now hampered further clinical development of ERCC1 as a first-in-class platinum biomarker, most notably in a prospective international, randomized Phase III clinical trial where there was an absence of clinical benefit for patients with low ERCC1 receiving a platinum agent [
8]. However, these mixed results have been largely attributed to problems pertaining to accurate detection of functional ERCC1/XPF (e.g., antibody specificity and splice variant expression), rather than its usefulness as a potential biomarker for response to platinum-based chemotherapy [
9]. Clear evidence from extensive data from biochemical, in vitro, and in vivo approaches suggests critical roles for the complex in repair of platinum-induced DNA damage, suggesting the inhibition of ERCC1/XPF as a potential means of sensitizing tumors to platinum-based chemotherapy.
ERCC1/XPF plays key roles in nucleotide excision repair, ICL repair, and homologous recombination. In the response to DNA-ICLs, ERCC1/XPF plays a key role in incising 5′ to the DNA lesion resulting in the unhooking of the interstrand crosslink from the DNA helix [
5]. In both replication-independent and -dependent pathways, ERCC1/XPF-mediated incision appears to be a generally required step for at least a subset of ICLs. In replication-dependent repair of interstrand crosslinks, however, several groups have also postulated that ERCC1/XPF nuclease activity is also essential at a second step downstream from initial unhooking likely during homologous recombination [
10]. In line with critical roles for ERCC1/XPF in ICL repair, we have previously shown that siRNA knockdown of ERCC1/XPF could enhance sensitivity of lung cancer cells to cisplatin [
3]. Additionally, this sensitivity was induced as a result of decreased DNA repair of interstrand and intrastrand crosslinks.
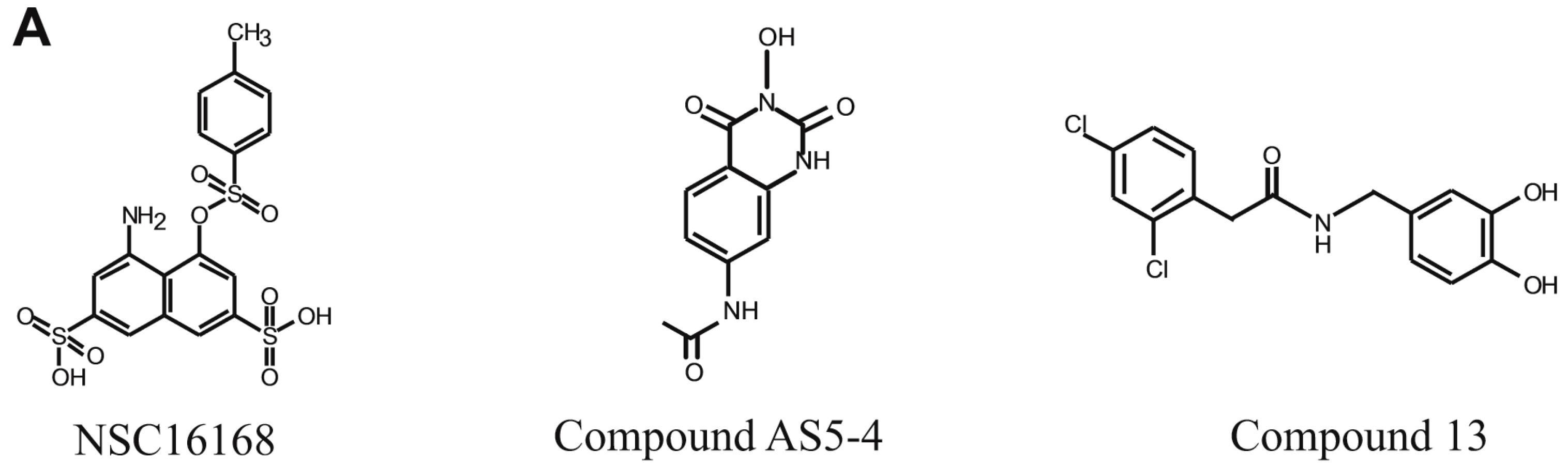
Several recent developments have been made in identifying small molecular inhibitors of ERCC1/XPF activity. The Melton and Saxty groups have published several papers identifying several classes of molecules capable of inhibiting ERCC1/XPF activity, including catechols, 3-hydroxypyridones, N-hydroxyimides, and hydroxypyrimidones, with micromolar potency in in vitro assays [
11,
12,
13]. In one of these studies, Compound 13 inhibited DNA repair in vitro, and led to increased γH2AX foci formation in human cells, suggesting inhibition of ERCC1/XPF in vitro [
12] (
Figure 1A). In another study, Compound AS5-4 disrupted ERCC1/XPF activity and enhanced cisplatin sensitivity in a melanoma cell line [
13] (
Figure 1A). Inhibitors of the ERCC1/XPF interaction have also been identified [
13]. An in silico screen identified the compound E-X PPI2 which could disrupt the ERCC1/XPF interaction, disrupt DNA repair mediated by ERCC1/XPF, and sensitize a melanoma cell line to cisplatin [
13]. In addition to these studies, we previously performed a high-throughput screen using the NCI-DTP (National Cancer Institute Developmental Therapeutics Program) diversity set and identified the compounds NSC16168 (
Figure 1A) and NSC143099, which were potent and selective inhibitors of ERCC1/XPF activity both in biochemical assays as well as in lung cancer cell lines [
14]. Furthermore, the compound NSC16168 was also capable of significantly enhancing tumor response to platinum-based chemotherapy in vivo [
14].
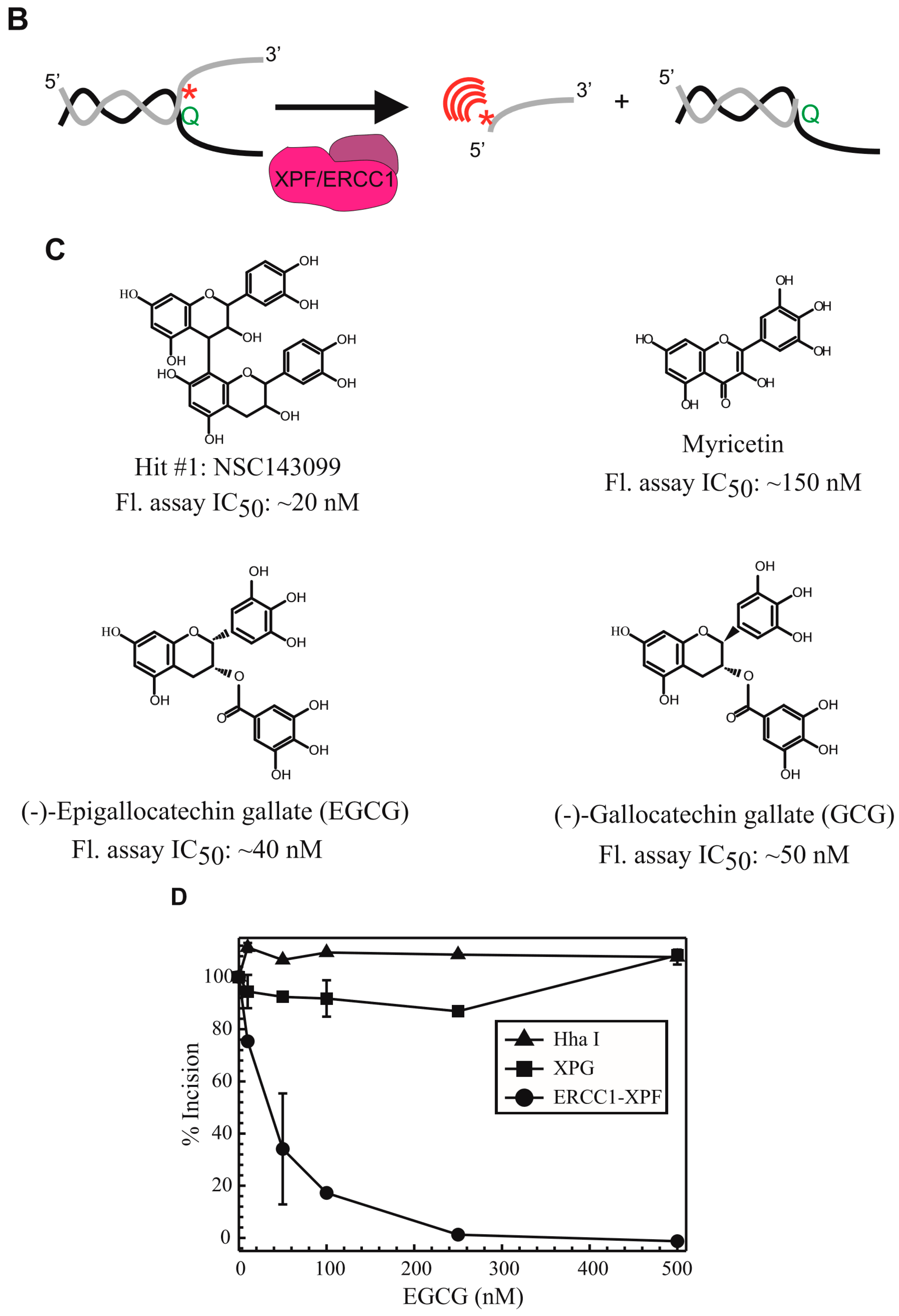
In this study, we expanded upon our previous identification of the lead compound NSC143099 as an ERCC1/XPF inhibitor by performing an in silico analysis to identify molecules with structural similarities to NSC143099 that may have activity against ERCC1/XPF both in vitro and in vivo. This preliminary screen and subsequent testing identified the green tea polyphenol epigallocatechin-3-gallate (EGCG), which has approximately 90% structural similarity to the partial structure of NSC143099, as a potent, partially reversible inhibitor of ERCC1/XPF activity in vitro. Interestingly, a number of previous studies have identified EGCG as a compound capable of enhancing cisplatin sensitivity in cancer cells and in xenograft models, but the mechanism of this interaction was not established [
15,
16,
17]. In addition, EGCG has been shown to pharmacologically inhibit 20S proteasome activity [
18,
19]. However, we are the first to identify the DNA endonuclease ERCC1/XPF as a pharmacological target of EGCG, helping to further explain previous observations in regard to EGCG and enhanced cisplatin sensitization. Further characterization of this compound shows it is capable of inhibiting ICL repair in vitro leading to enhanced sensitivity to cisplatin in lung cancer cell lines. Finally, we show that the inhibition of ERCC1/XPF by the EGCGprodrug, Pro-EGCG (EGCG octaacetate), could significantly enhance response to cisplatin in tumor xenografts in vivo by increasing tumor cell death and decreasing proliferation. Together, these data suggest EGCG or its prodrug may be a suitable candidate for further preclinical development as an agent capable of enhancing platinum sensitivity in tumors and overcoming drug resistance.
2. Materials and Methods
2.1. Cell Lines and Cell Culture
H1299 and H460 non-small cell lung cancer cell lines used in this study were cultured in RPMI-1640 medium (Dharmacon, Lafayette, CO, USA) supplemented with 10% Fetal Bovine Serum (Atlanta Biologicals, Flowery Branch, GA, USA) and 1% penicillin/streptomycin (Dharmacon, Lafayette). Cells were grown at 37 °C in 5% CO2. Cell lines were authenticated by the Biobanking and Correlative Sciences Core Facility at the Karmanos Cancer Institute.
2.2. In Vitro ERCC1/XPF Fluorescence Incision Assay
The in vitro ERCC1/XPF fluorescence incision assay was utilized to assess the ability of small molecules to inhibit ERCC1/XPF-mediated incision of a forked DNA substrate and was performed as previously described [
14]. Purified ERCC1/XPF and XPG (Xeroderma Pigmentosum Group G) was utilized for these experiments and the purification protocol is described in [
14]. The DNA substrate is described in Arora et al. [
14]. Briefly, the forked DNA substrate was designed to have a 14-base dsDNA region flanked by a 12-base non-complementary ssDNA region mimicking the ssDNA:dsDNA preferred substrate for ERCC1/XPF cleavage. One strand contained a site-specific fluorescein (F) modification (5′-GCCAGCGCTCGGAT (AminoC6dT) (FLSN) TTTTTTTTTTT-3′), whereas the complementary strand contained a site-specific DABCYL quencher (Q) (5′-TTTTTTTTTTT (AminoC6dT) (Dabcyl) ATCCGAGCGCTGGC-3′) directly opposed to the fluorescein modification. An uncleaved substrate thus would not release a fluorescent signal when excited at 485 nm due to the quenching activity of the DABCYL. On the other hand, ERCC1/XPF-mediated cleavage of the forked substrate would lead to the release of the fluorescein-labeled DNA into the solution and would result in an increased fluorescent signal when measuring emission at 525 nm. The substrate was also designed with a HhaI restriction enzyme cut site in the DNA duplex, which served as a positive control for these experiments. The reactions for this experiment consisted of 10 nM DNA annealed DNA duplex, 7.5 nM ERCC1/XPF or 7.5 nM XPG in buffer (50 mM Tris-HCl pH 8.0, 2 mM MgCl
2, 0.1 mM bovine serum albumin, 0.5 mM β-mercaptoethanol) or HhaI (20 U/mL) (New England Biolabs, Ipswich, MA, USA) and increasing concentrations of each compound prepared in DMSO. Reactions were incubated at 37 °C for 30 min after which samples were excited with a 485 nm laser and emission at 525 nm was measured using a Spectramax M5 plate reader (Molecular Devices, -San Jose, CA, USA). Data were plotted as percent fluorescence (% incision) relative to wells containing no compound.
2.3. Rapid Dilution Assay
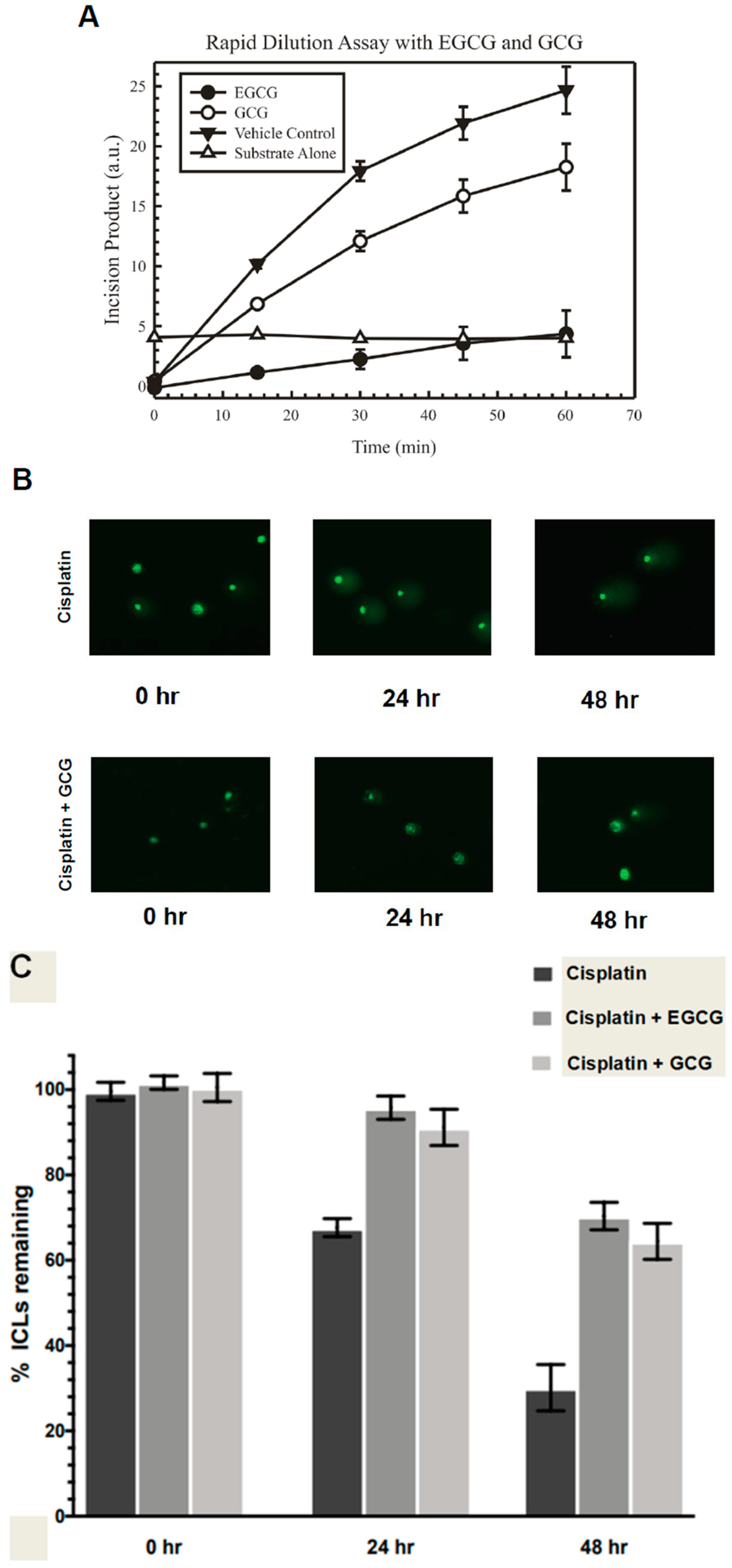
Rapid dilution assays were utilized to determine the irreversible or reversible binding of the EGCG and (-)-gallocatechin gallate compounds to ERCC1/XPF. These experiments were performed as described in Liu et al. [
20]. In a 10 μL reaction, the concentration of ERCC1/XPF enzyme was added at 100-fold higher than normal concentrations used in the fluorescence incision assay (7.5 nM) and then mixed with 10× the IC
90 concentration of either EGCG, (-)-gallocatechin gallate, or diluent control (90:10,
v/
v DMSO:glycerol) and incubated at 37 °C for 30 min. After incubation, 2 μL of the drug-enzyme solution was diluted in 198 μL consisting of reaction buffer and the fluorescent DNA forked substrate in a 96-well plate. The reaction was monitored and the fluorescence was measured at various time points over 60 min. Data from the experiment were represented as the increase in fluorescence over time indicating the amount of activity of ERCC1/XPF on the DNA substrate.
2.4. Modified Alkaline Comet Assay
Modified alkaline comet assays were utilized to assess interstrand crosslink repair and were performed essentially as previously described [
21,
22]. H460 cells were treated with 15 M EGCG or (-)-gallocatechin gallate for two hours and then cisplatin was added to the media using the IC90 concentration for the cell line used for an additional two hours. After treatment, cells were washed with PBS and complete media was added (24 h and 48 h samples) or immediately processed for analysis at 0 h post-treatment. Prior to cell harvesting but after the experimental treatment, cells were treated with 100 μM hydrogen peroxide (H
2O
2) for 15 min to induce DNA double strand breaks. Following treatment with hydrogen peroxide, cells were trypsinized, pelleted, resuspended, and counted. Approximately 10,000 cells were embedded in 1% low melting point agarose and added onto slides pre-coated with a layer of 1% normal melting point agarose and allowed to solidify. A top layer of 0.5% low melting point agarose was then added and allowed to solidify. Following solidification, slides were incubated for 1 h at 4 °C in the absence of light in lysis buffer (2.5 M NaCl, 10 mM Tris, 100 mM EDTA, 1% Triton X-100, pH 10). Slides were removed from the lysis buffer and excess buffer was removed. Slides were then placed in an electrophoresis tank containing 4 °C alkaline electrophoresis buffer (300 mM NaOH, 1 mM EDTA, pH > 13), incubated for 20 min followed by electrophoresis for 30 min at 0.7 V/cm, 300 mA. Slides were removed and placed for 10 min in neutralizing buffer (0.4 M Tris-HCl, pH 7.5). Slides were stained with SYBR green (Trevigen, Gaithersburg, MD, USA) and images were taken using a Nikon epifluorescence microscope at 20 magnification. For analysis, DNA tails for at least 50 cells were measured for each slide using Komet Assay Software 5.5F (Kinetic Imaging, Liverpool, UK). Data were analyzed and quantified as in Arora et al. [
14].
2.5. Clonogenic Survival Assays
Approximately 300–400 cells were seeded in triplicate in 60 mm dishes and allowed to attach for ~24 h. For treatments with EGCG or Pro-EGCG alone, the following day cells were titrated with the indicated concentrations of each drug for 4 h in serum-free medium followed by replacement with complete medium. For colony assays combining EGCG or Pro-EGCG with cisplatin, cells were pre-treated for 2 h and then cisplatin was added for 2 h (total treatment time with EGCG or Pro-EGCG was 4 h). After treatment, serum-free medium was replaced with complete medium. Cells were allowed to grow for approximately seven days after which plates were washed with PBS, fixed in 95% methanol, and stained in 20% ethanol containing 0.2% crystal violet dye. Colonies with >50 cells were counted using a light microscope, and percent colony survival was measured relative to the control for each group and normalized to 100%.
2.6. Chemicals
Cisplatin was purchased from Sigma-Aldrich (St. Louis, MO, USA) and prepared fresh prior to each experiment by making a 1 mM stock solution in PBS. (-)-epigallocatechin-3-gallate (EGCG) and (-)-gallocatechin gallate were purchased from Sigma-Aldrich and were prepared in dimethyl sulfoxide. Pro-EGCG was prepared as we previously described, and a 50 mM solution was prepared in DMSO for in vitro studies [
23].
2.7. In Vivo Studies
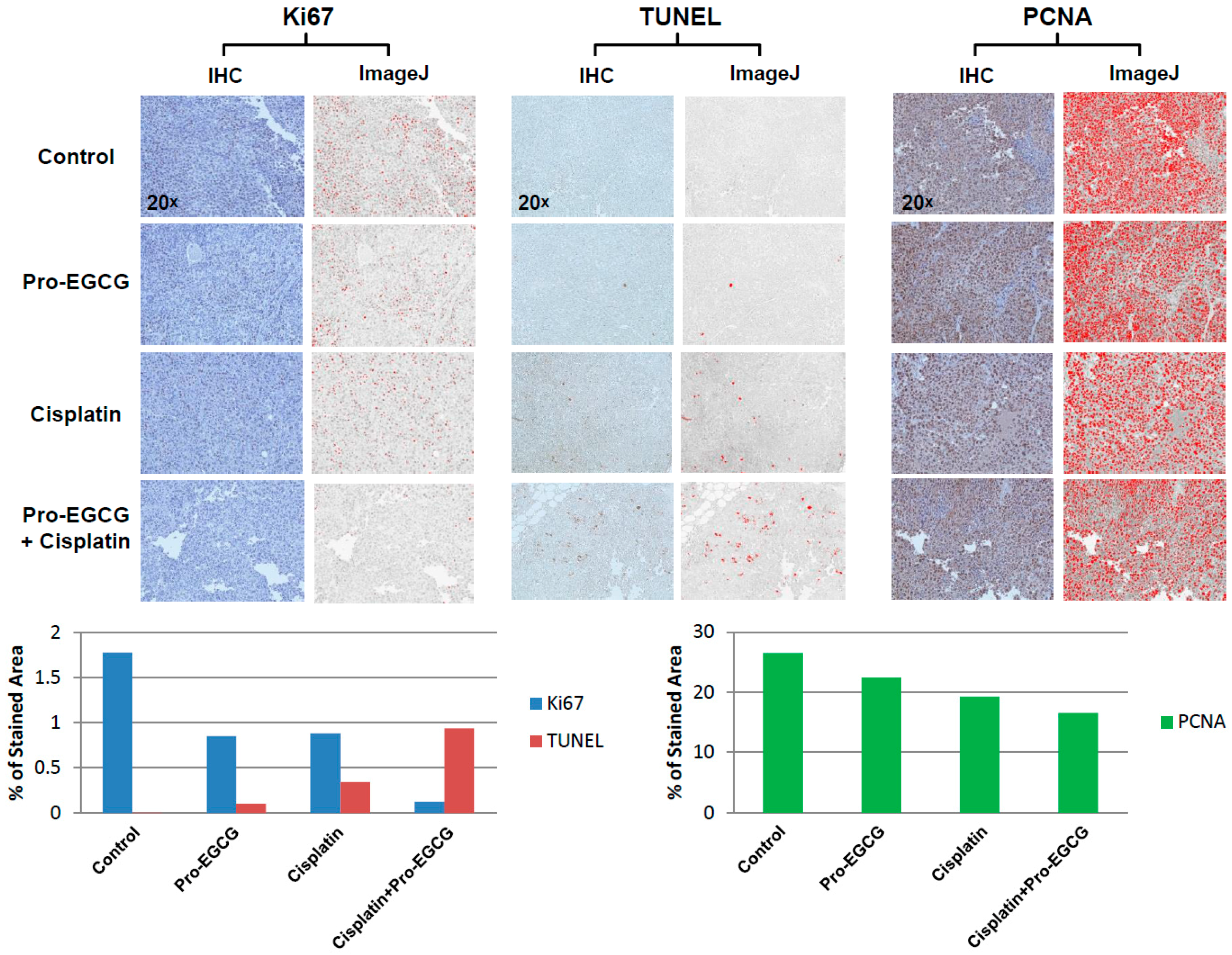
For the study, 20 female athymic nude mice (five mice per group) were purchased from Taconic Biosciences (Rensselaer, NY, USA) and were maintained in accordance with protocols approved by the Wayne State University Institutional Laboratory Animal Care and Use Committee. The mice were allowed to acclimate for 1 week. Approximately 2.5 × 106 H460 cells were suspended in 100 L RPMI media containing no fetal bovine serum or penicillin/streptomycin. Cells were injected subcutaneously into the right flank of each mouse. Tumor volume was measured by caliper measurements every day starting on day 3. Tumor volume was defined as (width2 × length/2). Weight of the mice was also measured regularly starting on the day of inoculation and through the experimental end-point. At three days post-inoculation, the mice were treated with vehicle or Pro-EGCG at 60 mg/kg by intraperitoneal IP injection. Pro-EGCG was prepared in DMSO and cremophor/ethanol (60:20:20 v/v/v). Once tumors reached a volume of ~100 mm3, cisplatin or vehicle treatment began. The mice were treated with 4 mg/kg pharmaceutical grade, sterile cisplatin three times weekly by IP injection. Drugs were prepared fresh daily. The mice were sacrificed once tumors reached ~1000 mm3 or at day 24, and the tumors were collected for further analysis of apoptosis and cell proliferation by immunohistochemistry.
2.8. Immunohistochemistry
Immunohistochemistry was performed by the Biobanking and Correlative Sciences Core at the Karmanos Cancer Institute (KCI). Subcutaneous tumors harvested from the mice were fixed in 10% formalin and were paraffin-embedded. Five μm slices were hematoxylin- and eosin-stained. Slides were stained using the Terminal nucleotidyl transferase-mediated nick end labeling assay (TUNEL) using an in situ apoptosis detection kit. Tissues were also probed for Ki67 and PCNA using standardized protocols optimized by the KCI Biobanking and Correlative Sciences Core. Images were taken at a magnification of 20. Images were quantified using ImageJ software and plotted as percent of stained area.
2.9. Statistical Analysis
The mouse xenograft study had five mice per treatment group, each bearing one tumor. Tumor volumes were log-transformed to meet the normality assumption, and growth curves were compared using a linear-mixed effects model with mice-specific effect as a random variable. P values were adjusted using Bonferroni correction. For the clonogenic survival assays, dose-response curve IC50 values for EGCG or Pro-EGCG in combination with cisplatin met distributional assumptions. Statistical comparisons were performed by two-sided unpaired t-test followed by Holm’s post-hoc analysis. Data analyses were completed using R version 3.5.0. and RStudio: Integrated Development for R (version 1.1.447, RStudio Inc.; Boston, MA, USA).
4. Discussion
Despite recent advances in cancer treatment such as using PD-1 and PD-L1 related therapies, platinum-based chemotherapy remains a mainstay option for a variety of tumor types either as first- or second-line therapy. Due to the prevalence of acquired or intrinsic resistance to platinum-based chemotherapy, it remains a valuable effort to identify factors that, when inhibited, can sensitize tumor cells to cisplatin. We previously have shown that ERCC1/XPF knockdown can sensitize ovarian and lung cancer cell lines to cisplatin [
3]. Additionally, our group previously identified the compound NSC16168 as a potent inhibitor of ERCC1/XPF that can sensitive tumors to cisplatin [
14]. The wide interest in ERCC1/XPF expression as a predictive biomarker for response to platinum-based chemotherapy also lends credence to the possibility of using inhibitors of this enzyme complex to enhance therapeutic response.
In our previous work, we performed a high-throughput screen and identified the compound NSC143099 as a potent inhibitor of ERCC1/XPF activity in vitro in both biochemical and cell-based assays [
14]. In this work, we performed an in silico screen to identify other agents with structural similarity to NSC143099 that could have the potential for inhibiting ERCC1/XPF activity. From this screen, we identified three hits with nanomolar potency against ERCC1/XPF activity in a fluorescence-based DNA-incision assay: myricetin, EGCG, and GCG (
Figure 1A). These compounds had substantial similarities and all shared a similar flavonoid structure. Myricetin was capable of inhibiting ERCC1/XPF activity with an IC
50 of ~150 nM in our DNA-incision assay. However, myricetin has also been shown to inhibit a variety of other enzymes, including MEK1, PI3Kγ, and the DNA endonuclease Ape1, among others [
28,
29,
30]. Due to the known targeting of this compound to Ape1, we did not move further with this compound in this study.
EGCG as an anti-cancer agent has been thoroughly investigated in multiple studies and in multiple cancer types, including in breast, colorectal, gastric, ovarian, and lung cancers [
31]. It has been established that EGCG has multiple cellular targets including work from the Dou lab, characterizing EGCG’s inhibitory effects on the proteasome [
27]. In line with these observations, treatment with EGCG or Pro-EGCG alone reduced clonogenicity in vitro in multiple lung cancer cell lines (
Figure 3B). Additionally, these effects appear to be independent of any toxicity induced by inhibition of ERCC1/XPF as the sensitivity of H1299 wild-type and ERCC1 knockout cells were identical to each other (
Figure 3C). Additionally, EGCG was described to have enhancing effects for cisplatin sensitivity both in vitro and in vivo. The studies assessing the cisplatin-sensitizing effects of EGCG implicated a number of different pathways and events as critical for this sensitization, including demethylation of gene promoters, increased expression of the copper transporter, CTR1, and enhancing autophagic flux [
15,
16,
17]. In this work, we identified another target of EGCG, ERCC1/XPF, which has important implications for understanding the mechanism of the EGCG-mediated sensitization of tumors to cisplatin and for improving current cancer treatment strategies.
The EGCG compound had no activity against XPG or HhaI, suggesting its inhibition is largely specific to ERCC1/XPF (
Figure 1B). Next, we showed that GCG, differing from EGCG only in the relative stereochemistry of the substitutions in the 2- and 3-positions, is a reversible inhibitor of ERCC1/XPF activity in a rapid dilution assay (
Figure 2B). Conversely, EGCG is either partially reversible with slow kinetics or is an irreversible inhibitor of ERCC1/XPF activity (
Figure 2A). This was similar to what we observed with another compound we identified from our original screen, NSC16168 [
14]. While there were differences in the reversibility of this inhibition in biochemical assays, both EGCG and GCG could inhibit repair of cisplatin-induced interstrand crosslinks in vitro (
Figure 2B,C). Not only did these compounds inhibit ERCC1/XPF activity in vitro, but this inhibition enhanced sensitivity to cisplatin in lung cancer cell lines (
Figure 3). Due to the poor bioavailability and the facile metabolic transformations of EGCG, we also utilized a prodrug form of EGCG for our studies. Previous study showed that Pro-EGCG was converted intracellularly into EGCG, presumably by cellular esterases. Furthermore, Pro-EGCG was better absorbed into the cells, giving higher accumulation of EGCG by at least 2.4-fold than when the cells were treated with similar levels of EGCG [
23,
27]. In our DNA-incision assay, as expected, we observed that Pro-EGCG has no inhibitory effect on the purified ERCC1/XPF protein (
Figure 3A). However, both EGCG and Pro-EGCG can sensitize lung cancer cells to an IC
50 dose of cisplatin in clonogenic assays indicating these cell line models have sufficient esterase activity to convert the Pro-EGCG to active EGCG (
Figure 3B).
Furthermore, after observing that EGCG could inhibit ERCC1/XPF and, along with Pro-EGCG, could sensitize lung cancer cells to cisplatin in vitro, we assessed the effects of combination treatment with cisplatin and Pro-EGCG in vivo. While Pro-EGCG and cisplatin had modest inhibitory effects on tumor growth as single agents, the combination treatment group had substantially smaller tumors at the day 24 endpoint (
Figure 4A,B). The tumors in the combination treatment group were barely palpable at the experimental endpoint. Further analysis of these tumors by immunohistochemistry revealed that this sensitization was associated with the decreased presence of markers of cell growth and DNA replication and a dramatic increase in TUNEL staining indicative of cell death (
Figure 5). Together these data suggest that EGCG and Pro-EGCG are potent inhibitors of ERCC1/XPF activity both in vitro and in vivo and that they are capable of sensitizing tumors to cisplatin therapy. In conclusion, we have identified the NSC143099 structural analogue, EGCG, as a potent inhibitor of ERCC1/XPF endonuclease activity capable of decreasing DNA repair in vitro and Pro-EGCG in enhancing cisplatin sensitivity in vivo. These data provide evidence that the green tea polyphenol, EGCG, and its prodrug could represent a potential structure for further pharmacological development in efforts to target the ERCC1/XPF endonuclease to enhance platinum-based chemotherapeutic response.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






